Идиопатический легочный фиброз (ИЛФ): современный подход к классификации и диагностике
Идиопатический легочный фиброз (ИЛФ) – это вариант идиопатической интерстициальной пневмонии (ИИП), характеризующийся неуклонным прогрессирующим течением и высокой смертностью. В отличие от большинства ИИП, иммуносупрессивная терапия не оказывает влияния на скорость прогрессирования ИЛФ. В течение последнего десятилетия установлена эффективность двух антифибротических препаратов в лечении ИЛФ – пирфенидона и нинтеданиба. Чтобы своевременно начать патогенетическую терапию, необходимо как можно быстрее установить диагноз ИЛФ на основании диагностического алгоритма, предполагающего анализ клинических, лабораторных и инструментальных данных, прежде всего результатов компьютерной томографии высокого разрешения (КТВР). При недостаточной информативности последней может быть использована малоинвазивная трансбронхиальная криобиопсия легкого, которая по точности сопоставима с хирургической биопсией легкого. Продолжается поиск молекулярно-биологических и генетических маркеров ИЛФ.
Согласно классификации Американского торакального общества/Европейского респираторного общества (ATS):1–112. /ERS):1–112. ) [1], идиопатический легочный фиброз (ИЛФ) представляет собой форму идиопатической интерстициальной пневмонии (ИИП) (табл. 1). Доля ИЛФ составляет 20-30% в структуре всех ИИП, а заболеваемость – от 7 до 17 случаев на 100 000 населения [2]. Мужчины болеют несколько чаще, чем женщины (соотношение мужчин/женщин приблизительно 1,5:1) [3]. ИЛФ развивается в основном у людей среднего и пожилого возраста: возраст 65% пациентов на момент постановки диагноза составляет 60 лет и более [4].
| Частые формы ИИП |
| Идиопатический легочный фиброз (ИЛФ) |
| Идиопатическая неспецифическая интерстициальная пневмония |
| Респираторный бронхиолит, ассоциированный с интерстициальным заболеванием легких |
| Десквамативная интерстициальная пневмония |
| Криптогенная организующая пневмония |
| Острая интерстициальная пневмония |
| Редкие формы ИИП |
| Идиопатическая лимфоцитарная интерстициальная пневмония |
| Идиопатический плевропаренхиматозный фиброэластоз |
| Неклассифицируемые формы (ИИП) |
В 2018 году P. Wolters и соавт. предложили выделять 4 варианта легочного фиброза в зависимости от патогенеза заболевания (табл. 2) [5]. ИЛФ характеризуется прогрессирующим течением с развитием дыхательной недостаточности и среди всех ИИП обладает самым неблагоприятным прогнозом: средняя выживаемость составляет от 2 до 5 лет [6,7]. Высокая смертность пациентов с ИЛФ объясняется особенностями патогенеза заболевания – преобладанием фиброза при незначительной выраженности воспалительных изменений [8,9]. Основным механизмом, приводящим к развитию прогрессирующего легочного фиброза, является персистирующее повреждение альвеолярного эпителия с последующим нарушением процессов его регенерации, избыточным отложением компонентов внеклеточного матрикса, активацией фибробластов и миофибробластов [10]. Указанные изменения определяют неэффективность традиционной иммуносупрессивной терапии у пациентов с ИЛФ [11]. Тем не менее, в настоящее время достигнуты значительные успехи в лечении ИЛФ, связанные с применением антифибротических препаратов – пирфенидона (антагониста трансформирующего фактора роста бета – TGF β) и нинтеданиба (множественного ингибитора тирозинкиназ), замедляющих уменьшение легочных объемов, в первую очередь, форсированной жизненной емкости легких (ФЖЕЛ), и улучшающих выживаемость без прогрессирования заболевания [12]. При отсутствии противопоказаний трансплантация легких также рассматривается в качестве варианта лечения у пациентов с прогрессирующим ИЛФ, осложнившимся тяжелой дыхательной недостаточностью [13,14].
| Группа 1: ЛФ, индуцированный дисфункцией эпителиальных клеток | ИЛФ |
| Группа 2: ЛФ, индуцированный дисфункцией клеток воспалени | Системная склеродермия, ревматоидный артрит, синдром Шегрена, экзогенный аллергический альвеолит, саркоидоз, НСИП |
| Группа 3: ЛФ, вызванный приемом лекарственных препаратов или воздействием профессиональных факторов | Асбестоз, силикоз, лекарственное поражение легких |
| Группа 4: ЛФ, связанный с курением | Десквамативная интерстициальная пневмония, респираторный бронхиолит, ассоциированный с интерстициальным заболеванием легких, Лангерганс-клеточный гистиоцитоз |
Клиническая картина
Основные жалобы у пациентов с ИЛФ – прогрессирующая одышка и сухой кашель, усиливающиеся при физической нагрузке. Реже отмечаются боль и дискомфорт в грудной клетке, повышенная утомляемость, общая слабость, снижение массы тела. В ряде случаев заболевание на начальных этапах протекает бессимптомно, а первыми проявлениями оказываются изменения функциональных легочных параметров [1]. Типичным аускультативным феноменом при ИЛФ является крепитация, преимущественно в задне-базальных отделах легких. У больных c развернутой стадией ИЛФ могут отмечаться признаки вторичной артериальной легочной гипертензии с развитием легочного сердца и правожелудочковой сердечной недостаточности [15].
При ИЛФ может определяться незначительное повышение СОЭ. Несмотря на наличие прогрессирующей дыхательной недостаточности, выраженное увеличение концентрации гемоглобина наблюдается крайне редко. уменьшением всех легочных объемов в сочетании со снижением диффузионной способности легких (DLCO). Одним из ранних проявлений ИЛФ может быть изолированное снижение DLCO при относительной сохранности легочных объемов. Также к ранним проявлениям ИЛФ относят увеличение альвеолоартериального градиента по кислороду, что часто характеризуется нормальными показателями сатурации крови в покое и десатурацией при физической нагрузке [16].
Диагностический алгоритм
Диагноз ИЛФ основывается на отсутствии известных причин легочного фиброза и наличии картины обычной интерстициальной пневмонии (ОИП) [17]. Даже при наличии гистологической картины ОИП при хирургической биопсии легкого (ХБЛ) окончательный диагноз требует исключения других патологических состояний, ассоциированных с развитием ОИП, включая диффузные заболевания соединительной ткани, пневмокониозы, поражение легких, связанное с приемом лекарственных препаратов, семейный легочный фиброз [18]. При отсутствии данных за альтернативный диагноз, согласно действующим клиническим рекомендациям [4], диагноз ИЛФ устанавливают на основании характерных данных компьютерной томографии высокого разрешения (КТВР) и, при необходимости, результатов биопсии легкого (табл. 3). Следует отметить, что в представленной гистологической классификации выделены «возможный ИЛФ» и «вероятный ИЛФ», когда невозможно однозначно подтвердить или исключить наличие ИЛФ. В таком случае показана повторная оценка данных КТВР и биопсии легкого для уточнения диагноза.
| КТ-картина | Гистологические данные | Диагноз |
|---|---|---|
| ОИП | ОИП | ИЛФ |
| Вероятная ОИП | ||
| Возможная ОИП | ||
| Неклассифицируемый фиброз | ||
| Не соответствует ОИП | Не-ИЛФ | |
| Возможная ОИП | ОИП Вероятная ОИП | ИЛФ |
| Возможная ОИП | Вероятный ИЛФ | |
| Неклассифицируемый фиброз | ||
| Не соответствует ОИП | Не-ИЛФ | |
| Не соответствует ОИП | ОИП | Возможный ИЛФ |
| Вероятная ОИП | Не-ИЛФ | |
| Возможная ОИП | ||
| Неклассифицируемый фиброз | ||
| Не соответствует ОИП |
КТ-диагностика
КТВР играет ключевую роль в диагностике ИЛФ и позволяет установить диагноз приблизительно в 2/3 случаев. В ряде исследований было показано, что КТ-картина типичной ОИП по данным КТВР согласуется с наличием гистологической картины типичной ОИП по данным биопсии легкого в 90-100% случаев [4]. Наличие достоверных КТ-признаков ОИП в настоящее время считают достаточным для диагностики ИЛФ без биопсии легкого. Проведение хирургической биопсии легкого (ХБЛ) рекомендуется при наличии КТ-картины, не типичной для ОИП. В таких случаях диагноз устанавливают на основании сочетания данных КТВР и гистологической картины (табл. 3). Таким образом, точная интерпретация данных КТВР является необходимым условием для постановки диагноза [1].
В настоящее время выделяют три КТ-варианта ОИП «типичная ОИП», которая исключает необходимость проведения ХБЛ, «возможная ОИП» и «не соответствует ОИП». При наличии последних двух вариантов требуется проведение ХБЛ [19].
КТ-картина типичной ОИП включает в себя преимущественно базальные и периферические ретикулярные изменения с образованием сотового легкого в сочетании с тракционными бронхоэктазами или без них. Критериями «сотового легкого» считают преимущественно субплевральные кисты диаметром 3-10 мм с четкими, относительно толстыми стенками (1-3 мм), расположенные слоями. Все КТ-признаки, рассматриваемые как «не соответствующие» ОИП, должны отсутствовать (рис. 1). Если все вышеуказанные критерии выполнены, данные КТВР достаточны для диагностики ОИП, а необходимости в проведения биопсии легкого нет [4]. Относительно признаков типичной ОИП заключения разных специалистов обычно хорошо согласуются [20,21]. Тем не менее, следует отметить, что ОИП и ИЛФ не являются синонимами, так как КТизменения, характерные для ОИП, могут отмечаться при ряде других заболеваний, прежде всего диффузных заболеваниях соединительной ткани.
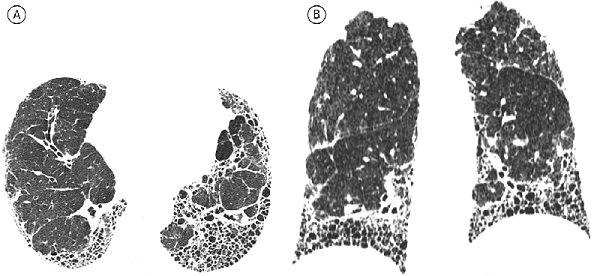 Рис. 1. КТ-картина типичной ОИП у женщины 77 лет. (А) На аксиальном срезе определяются ретикулярные изменения, тракционные брон-хоэктазы, зоны «сотового легкого». (В) При корональной реконструкции отмечается апикобазальный градиент поражения легочной ткани [22].
Рис. 1. КТ-картина типичной ОИП у женщины 77 лет. (А) На аксиальном срезе определяются ретикулярные изменения, тракционные брон-хоэктазы, зоны «сотового легкого». (В) При корональной реконструкции отмечается апикобазальный градиент поражения легочной ткани [22].
При возможной ОИП наблюдаются преимущественно базальные и периферические ретикулярные изменения без формирования зон сотового легкого. При этом изменения, не соответствующие ОИП, отсутствуют (рис. 2). Картина возможной ОИП менее специфична для ИЛФ, чем картина типичной ОИП. В данном случае дифференциальный диагноз следует проводить, в первую очередь, с неспецифической интерстициальной пневмонией (НСИП), для которой характерны отсутствие участков сотового легкого, преобладание затемнений по типу «матового стекла» над ретикулярными изменениями, относительная сохранность субплевральных зон. Участки сотовой трансформации редко встречаются при НСИП. В одном исследовании они были выявлены менее чем у 5% пациентов с идиопатической НСИП [23].
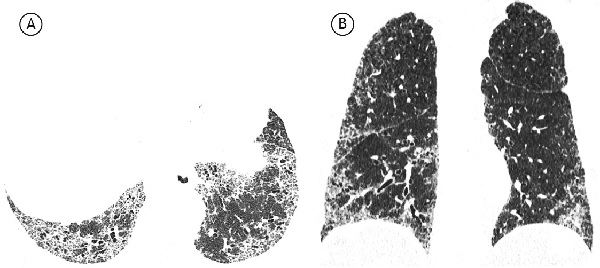 Рис. 2. КТ-картина возможной ОИП у мужчины 75 лет. (А) На аксиальном срезе определяются ретикулярные изменения, тракционные брон-хоэктазы, зоны «сотового легкого». (В) При корональной реконструкции отмечается апикобазальный градиент поражения легочной ткани [22].
Рис. 2. КТ-картина возможной ОИП у мужчины 75 лет. (А) На аксиальном срезе определяются ретикулярные изменения, тракционные брон-хоэктазы, зоны «сотового легкого». (В) При корональной реконструкции отмечается апикобазальный градиент поражения легочной ткани [22].
Изменения по данным КТВР, которые считают не соответствующими ОИП, включают в себя следующие: а) преобладание изменений в верхних и средних отделах легких; б) преимущественно перибронховаскулярные изменения; в) значительные по размеру зоны затемнения по типу «матового стекла», распростра ненность которых превышает таковую ретикулярных изменений; г) двусторонние очаговые изменения, преимущественно в верхних отделах легких; д) наличие кист (множественных, двусторонних) вне зон фиброза; е) картина мозаичного затемнения легочной ткани/ наличие «воздушных ловушек» (двусторонние изменения в трех и более долях); ж) наличие зон консолидации (рис. 3).
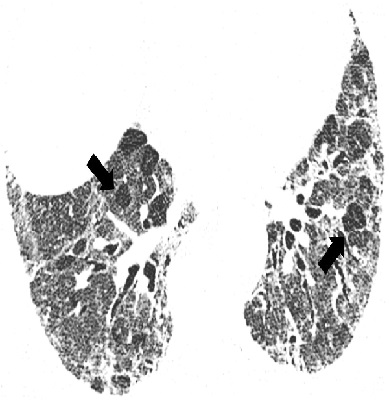 Рис.3. КТ-картина, не соответствующая ОИП, у пациентки 61 года с хроническим экзогенным аллергическим аль веолитом. На аксиальном срезе выявляются ретикулярные изменения в сочетании с мозаичным распространением зон затемнения по типу «матового стекла» [22]
Рис.3. КТ-картина, не соответствующая ОИП, у пациентки 61 года с хроническим экзогенным аллергическим аль веолитом. На аксиальном срезе выявляются ретикулярные изменения в сочетании с мозаичным распространением зон затемнения по типу «матового стекла» [22]
Несмотря на высокую вероятность наличия ИЛФ при типичной ОИП по данным КТВР, отсутствие зарактерной КТ-картины не должно служить основанием для исключения диагноза ИЛФ [22]. В 2017 г. D. Lynch и соавт. предложили новую КТ-классификацию ОИП, в которой впервые выделена группа неопределенной ОИП (табл. 4) [24].
| Типичная ОИП | Вероятная ОИП | Неопределенная ОИП | Наименее вероятно соответствует ОИП |
|---|---|---|---|
| Преобладание в базальных и субплевральных отделах (редко диффузные изменения); часто неоднородное распределение Зоны «сотового легкого»; ретикулярные изменения с периферическими тракционными бронхо эктазами и бронхиолоэктазами; отсутствие данных за альтернативный диагноз | Преобладание в базальных и субплевральных отделах; часто неоднородное распределение Ретикулярные изменения с периферическими тракционным бронхоэктазами и бронхиолоэктазами; отсутствие зон «сотового легкого»; отсутствие данных за альтернативный диагноз | Вариабельное или диффузное распределение Наличие фиброза в сочетании с небольшими по объему изменениями, не соответствующими ОИП | Преобладание в верхних и средних отделах легких; перибронховаскулярное распределение с относительной сохранностью субплевральных зон Любое из нижеперечисленного: преобладание зон консолидации; значительные по размеру зоны затемнения по типу «матового стекла» (при отсутствии обострения ИЛФ); диффузные очаговые или кистозные изменения; выраженное мозаичное затемнение легочной ткани с наличием «воздушных ловушек» |
Клиническое течение ИЛФ может быть различным. У большинства пациентов отмечается медленно прогрессирующее течение, однако у некоторых пациентов происходит стабилизация патологического процесса, тогда как у других отмечается довольно быстрое прогрессирование заболевания. Что касается выраженности легочных изменений по данным КТВР, то зоны затемнения по типу «матового стекла» чаще всего трансформируются в ретикулярные изменения, которые, в свою очередь, могут прогрессировать и формировать зоны «сотового легкого», размер которых со временем обычно увеличивается. Следует отметить, что общий паттерн легочных изменений также может изменяться: так, КТкартина возможной ОИП может трансформироваться в типичную ОИП [22].
Биопсия легкого
Если однозначные данные о наличии ИЛФ при КТВР отсутствуют, то для подтверждения диагноза показано выполнение хирургической биопсии легких, которую чаще проводят с помощью видеоторакоскопической методики. С целью повышение эффективности биопсия легких должна производиться из разных долей легких. Хотя ХБЛ является наиболее достоверным методом определения гистологической картины ИИП, ее проведение связано с риском возникновения ряда осложнений, наиболее тяжелым из которых является обострение ИЛФ, особенно у пациентов с тяжелой дыхательной и/или сердечной недостаточностью [25]. В связи с этим решение о ее проведении должно приниматься индивидуально с учетом клинической картины, возможных преимуществ для постановки точного диагноза, а также согласия пациента.
В течение последнего десятилетия для гистологического подтверждения диагноза ИЛФ и других вариантов ИИП разработана методика трансбронхиальной криобиопсии легкого (ТБКБЛ). Ее основными преимуществами являются малоинвазивность, отсутствие необходимости в проведении интубации и ингаляционного наркоза и, вследствие этого, низкая частота развития осложнений в сочетании с возможностью получения большого по объему биоптата легкого, достаточного, в абсолютном большинстве случаев, для гистологической верификации диагноза [26]. Так, у пациентов без типичной картины ОИП по данным КТВР проведение ТБКБЛ позволяло установить диагноз приблизительно в 2/3 случаев, что сопоставимо с эффективностью ХБЛ в сходной ситуации [27]. При этом для ТБКБЛ характерны более низкий риск периоперационных осложнений (чаще всего отмечают развитие пневмоторакса и не угрожающего жизни кровотечения в месте проведения биопсии) и смерти, более короткий период госпитализации, что позволяет проводить ТБКБЛ у пациентов с высоким уровнем анестезиологического риска и наличием противопоказаний к ХБЛ [28].Таким образом, внедрение ТБКБЛ в клиническую практику может расширить показания к биопсии легкого и повысить диагностическую точность алгоритма обследования пациентов с подозрением на ИЛФ.
При морфологическом исследовании у пациентов с подозрением на ИЛФ G. Raghu и соавт. выделяют пять возможных гистологических паттернов заболевания (табл. 5) [29,4]. В сочетании с рентгенологическими данными они используются для подтверждения/исключения диагноза ИЛФ (табл. 3) [4,30].
Дифференциальный диагноз
У пациентов с подозрением на ИЛФ должен проводиться тщательный дифференциальный диагноз. При выявлении КТ-картины, соответствующей вероятной или возможной ОИП, что происходит довольно часто, в круг дифференциального диагноза следует включать, в первую очередь, хронический экзогенный аллергический альвеолит и фибротический вариант НСИП. Тем не менее, у части пациентов рекомендованная в данном случае ХБЛ не проводится в связи с наличием противопоказаний (тяжелой дыхательной недостаточности, сопутствующих заболеваний, возрастных ограничений) или нежеланием пациента.
При проведении дифференциального диагноза важно также исключить поражение легких в рамках системного заболевания соединительной ткани, в частности, ревматоидного артрита, системной склеродермии, дерматомиозита, синдрома Шегрена [31], в том числе при наличии КТ-картины типичной ОИП. При наличии у пациента отдельных клинических проявлений или повышения уровня лабораторных аутоиммунных маркеров, не соответствующих конкретному системному заболеванию соединительной ткани, может быть установлен диагноз интерстициальной пневмонии с аутоиммунными чертами [32].
Генетические маркеры ИЛФ
В настоящее время выявлен ряд мутаций и полиморфизмов генов, участвующих в ремоделировании легочной ткани и регуляции врожденного и приобретенного иммунитета, ассоциированных с развитием ИЛФ [33]. К ним относятся, в частности, мутации в генах, кодирующих сурфактантные протеины А и D (S):1–112. P-A и S):1–112. PD), описанные при семейных формах ИЛФ [34]. В ряде исследований выявлена ассоциация генетических полиморфизмов с прогнозом заболевания: в частности, наличие отдельных однонуклеотидных полиморфизмов в гене TLR-3 (Toll-подобный рецептор 3-го типа) ассоциировано с более быстрым прогрессированием заболевания [35]. Также при ИЛФ описан ряд полиморфизмов в генах муцина 5B (MUC5B) и TOLLIP (протеин, взаимодействующий с Toll-подобным рецептором) [36]. Хотя исследование генетических полиморфизмов не является частью диагностического алгоритма при ИЛФ, продолжается поиск генетических маркеров, способных служить предикторами различных вариантов течения заболевания и ответа на терапию.
Обострение ИЛФ
Обострение ИЛФ – это тяжелое жизнеугрожающее состояние, проявляющееся в виде быстрого нарастания дыхательной недостаточности у пациентов с ранее установленным диагнозом ИЛФ [37]. Как правило, характеризуется крайне тяжелым течением; смертность в ряде исследований достигала 85% [38]. В отличие от стабильного или медленно прогрессирующего течения ИЛФ, критерии диагностики его обострения определены менее четко. Согласно данным Н. Collard и соавт. [39], критерии обострения ИЛФ включают в себя наличие предшествующего или впервые выявленного ИЛФ с резким нарастанием одышки, развитием дыхательной недостаточности за предшествующие 30 дней без установленной причины, а также появление новых зон затемнения легочной ткани по типу «матового стекла» и/или консолидации на фоне имевшихся ранее изменений, соответствующих ОИП – зон ретикулярных изменений и «сотового легкого» (рис. 4) [40]. Тем не менее, вышеуказанные критерии обладают низкой специфичностью, в связи с чем при подозрении на обострение ИЛФ должен проводиться дифференциальный диагноз с инфекционным процессом, тромбоэмболией легочной артерии и ее ветвей, пневмотораксом, а также острой левожелудочковой недостаточностью с развитием отека легких [41].
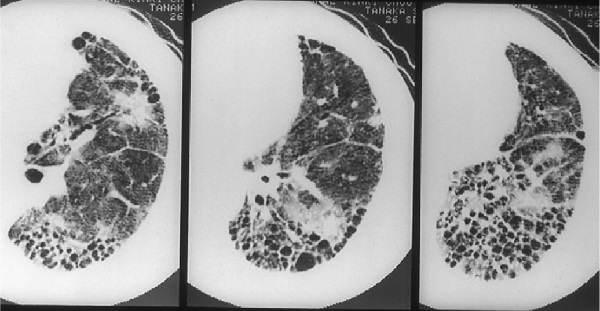 Рис. 4. Мультифокальный вариант обострения ИЛФ. По данным КТВР определяются затемнения легочной ткани по типу «матового стекла» и консолидации в центральных и периферических отделах легкого в сочетании с субплевральными изменениями по типу «сотового легкого»[40]
Рис. 4. Мультифокальный вариант обострения ИЛФ. По данным КТВР определяются затемнения легочной ткани по типу «матового стекла» и консолидации в центральных и периферических отделах легкого в сочетании с субплевральными изменениями по типу «сотового легкого»[40]
Заключение
Появление новых методов лечения, в частности, антифибротических препаратов, и неэффективность традиционной иммуносупрессивной терапии при ИЛФ подчеркивают важность как можно более ранней постановки диагноза и начала терапии. В течение последнего десятилетия был достигнут значительный прогресс в разработке диагностических алгоритмов для пациентов с ИЛФ. Этому способствовало повышение качества визуализационных методов, более полное понимание роли биопсии легких и разработка гистологических критериев ИЛФ. Все вышеперечисленные параметры должны исследоваться мультидисциплинарной командой специалистов, что в настоящий момент является стандартом диагностики ИЛФ. Несмотря на достигнутые успехи, в диагностике ИЛФ остаются нерешенные вопросы, в основном касающиеся применения инвазивных методов диагностики, в частности, хирургической биопсии легкого. Необходимо продолжать поиск молекулярно-биологических и генетических маркеров ИЛФ и разработку малоинвазивных биопсийных методов для максимально раннего установления диагноза, определения прогноза и разработки стратегии терапии ИЛФ.
Идиопатические интерстициальные пневмонии
Рассмотрены интерстициальные заболеваний легких (ИЗЛ) с точки зрения классификации и терминологии, а также клинические формы и диагностические критерии идиопатических интерстициальных пневмоний (ИИП), идиопатического легочного фиброза и других ИЗЛ. Рассмо
Interstitial pulmonary diseases (IPD) were considered from the point of view of classification and terminology, and also clinical forms and diagnostic criteria of idiopathic interstitial pneumonia (IIP), idiopathic pulmonary fibrosis and other IPD. Approaches to the IIP treatment in patients were examined.
В группу интерстициальных заболеваний легких (ИЗЛ) включают несколько десятков отдельных нозологических форм, отличающихся по этиологии, особенностям патогенеза и морфологической картине, имеющих различную клинику и прогноз. Терминологические и классификационные подходы к этим заболеваниям неоднократно менялись, дополнительно затрудняя и без того непростую диагностическую работу с данной категорией пациентов. Даже сегодня, несмотря на наличие общепринятой классификации ИЗЛ, термин «интерстициальная пневмония» ассоциируется у врача скорее с вирусной инфекцией, чем с заболеванием, требующим обязательной морфологической верификации и имеющим довольно серьезный прогноз [1].
Напомним основные определения.
Пневмония — группа острых инфекционных (преимущественно бактериальных) заболеваний, характеризующихся очаговым поражением респираторных отделов легких с обязательным наличием внутриальвеолярной экссудации. Из рубрики «пневмония» исключены заболевания, вызванные физическими (лучевой пневмонит) или химическими («бензиновая» пневмония) факторами, а также имеющими аллергическое («эозинофильная пневмония») или сосудистое (инфаркт легкого вследствие ТЭЛА) происхождение. Воспалительные процессы в легких при кори, краснухе, гриппе и др. рассматриваются не в рубрике «пневмония», а в рамках соответствующих нозологических форм [2].
Пневмонит (альвеолит) — воспалительный процесс, часто иммунного, неинфекционного характера, затрагивающий преимущественно паренхиматозный интерстиций (альвеолярные стенки) и экстраальвеолярную соединительную ткань легких без обязательной внутриальвеолярной экссудации. Ряд авторов разграничивает понятия «пневмонит» и «альвеолит», предполагая, что при альвеолите воспалительный процесс локализуется преимущественно в альвеолах, а при пневмоните воспаление затрагивает и другие структуры паренхимы легких, однако практического значения такое разделение не имеет, и термины часто используются как синонимы [3].
Термин «пневмонит» отражает не конкретную нозологическую форму, а особенности патологического процесса. Поражение легких по типу пневмонита (альвеолита) может развиваться при самых разных заболеваниях: идиопатических интерстициальных пневмониях, лекарственных поражениях легких, системных заболеваниях соединительной ткани, гиперчувствительном пневмоните, саркоидозе и др. Следует учесть, что при каждом из этих заболеваний пневмонит является обязательным, но далеко не единственным проявлением поражения легких. Причины, клинические проявления, направления лечения и прогноз при этих заболеваниях различны, поэтому при выявлении признаков пневмонита такое большое значение имеют морфологическая верификация и дальнейшая нозологическая диагностика.
С современных позиций ИЗЛ представляют собой гетерогенную группу заболеваний, общими чертами которых являются поражение интерстициальной ткани легких по типу продуктивного пневмонита с последующим формированием фиброза, прогрессирующая одышка при нагрузке, непродуктивный кашель, крепитация, диффузные изменения при рентгенографии и компьютерной томографии легких, рестриктивные вентиляционные нарушения, снижение диффузионной способности легких и нарастающая дыхательная недостаточность [4].
В настоящее время в большинстве стран, в том числе и в России, используется классификация ИЗЛ, принятая Согласительной комиссии Американского торакального общества и Европейского респираторного общества (ATS/ERS, 2002 г.) [5]. Согласно этой классификации выделяют четыре группы ИЗЛ: ИЗЛ известной этиологии, гранулематозы, идиопатические интерстициальные пневмонии, другие ИЗЛ (рис.).
.gif)
Идиопатические интерстициальные пневмонии (ИИП) — типичные представители группы ИЗЛ неизвестной этиологии, имеющие много сходных клинических, рентгенологических и функциональных признаков, но принципиально разную морфологическую картину, обусловливающую особенности клиники, ответ на терапию и прогноз. Принципом построения клинико-патологической классификации ATS/ERS является соответствие каждой клинической форме ИИП определенного гистологического варианта ИИП (табл. 1).
Клинические особенности больных с различными видами ИИП приведены в табл. 2.
_575.gif)
Идиопатический легочный фиброз
Идиопатический легочный фиброз (ИЛФ) является одним из наиболее часто встречающихся заболеваний из группы ИИП. Синонимом ИЛФ являются «идиопатический фиброзирующий альвеолит» — термин, традиционно используемый в нашей стране.
Заболевание чаще всего встречается у пациентов в возрасте старше 50 лет. Основными жалобами больных являются нарастающая одышка и непродуктивный кашель. Начало болезни, как правило, незаметное, болезнь прогрессирует довольно медленно, пациенты успевают адаптироваться к своей одышке и на момент обращения имеют анамнез заболевания длительностью до 1–3 лет. Лихорадка и кровохарканье для больных ИФЛ не характерны. Другими симптомами могут быть общая слабость, артралгии, миалгии, изменение ногтевых фаланг в виде «барабанных палочек». Типичным аускультативным феноменом при ИЛФ является инспираторная крепитация, которую сравнивают с «треском целлофана». По мере прогрессирования заболевания появляются признаки дыхательной недостаточности и легочного сердца, снижение массы тела вплоть до кахексии. Данные лабораторного обследования неспецифичны. ИЛФ относится к рестриктивным легочным заболеваниям, поэтому характерными функциональными особенностями заболевания является снижение статических легочных объемов, выявляемое при бодиплетизмографии. Одним из ранних признаков заболевания является снижение DLCO. Спирометрический показатель FEV1/FVC находится в пределах нормы или повышен.
Наиболее частыми рентгенографическими признаками ИЛФ являются двусторонние изменения ретикулярного характера, более выраженные в нижних отделах легких. На ранних этапах развития заболевания может наблюдаться лишь некоторое уменьшение объема легочных полей и понижение прозрачности легких по типу «матового стекла». При прогрессировании заболевания ретикулярный паттерн становится более грубым, тяжистым, появляются округлые кистозные просветления, отражающие формирование «сотового легкого». Для уточнения рентгенологической картины целесообразно проведение мультиспиральной компьютерной томографии органов грудной клетки.
Поскольку возможности диагностики ИИП ограничены, а данные обследования не всегда специфичны, «золотым» диагностическим стандартом всех ИИП является биопсия легких: открытая либо торакоскопическая. Особая необходимость в выполнении биопсии возникает в случаях наличия не вполне типичной клинической и/или рентгенологической картины, возрасте пациента менее 50 лет, наличии системных признаков заболевании, быстром прогрессировании заболевания. Необходимым условием является преобладание пользы от постановки правильного диагноза над риском хирургической манипуляции.
Существует диагностический подход, позволяющий с большой вероятностью установить диагноз ИЛФ в тех случаях, когда проведении биопсии невозможно. Для этого необходимо, чтобы у пациента имелись четыре из четырех больших критериев и хотя бы три из четырех малых критериев.
Большие критерии
Малые критерии
Современная терапия ИЛФ построена, в основном, на противовоспалительной терапии (кортикостероиды и цитостатики (ЦС)), т. е. препаратах, способных воздействовать на воспалительные и иммунологические звенья развития заболевания. Базой такого подхода служит положение, что хроническое воспаление предшествует и неизбежно ведет к фиброзу и что агрессивное подавление воспаления может блокировать последующее формирование фиброзных изменений.
Широко используются три режима противовоспалительной терапии: монотерапия глюкокортикостероидами (ГКС), комбинация ГКС с азатиоприном и комбинация ГКС с Циклофосфаном. ATS/ERS рекомендует комбинированные режимы как более предпочтительные [6]. Терапия проводится, как минимум, в течение 6 месяцев. Обязательно тщательное мониторирование побочных эффектов терапии. При назначении цитостатиков мониторинг больных должен включать общий анализ крови еженедельно в течение первого месяца, затем один раз каждые 2–4 недели; при терапии Циклофосфаном требуется еженедельный анализ мочи на гематурию.
В случае выбора монотерапии ГКС начальная суточная доза преднизолона составляет 1 мг/кг идеального веса в сутки (максимум до 80 мг/сут). Через 4 недели проводится оценка переносимости такой терапии. Если произошло улучшение или стабилизация функциональных показателей, то в течение последующих 3 месяцев суточную дозу преднизолона уменьшают. При отсутствии ответа на стероиды добавляют азатиоприн [7].
Альтернативным подходом, сфокусированным на снижении избыточной депозиции матрикса в легких или ускорении распада коллагена, является антифибротическая терапия. К числу антифиброзных препаратов относятся D-пеницилламин, колхицин, интерферон гамма-1 b, пирфенидон.
Доказано повышение эффективности терапии при добавлении к противовоспалительным препаратам N-ацетилцистеина в дозе 600 мг 3 раза в сутки. В настоящее время ведущие эксперты при лечении ИЛФ отдают предпочтение схеме, включающей преднизолон, азатиоприн и N-ацетилцистеин [8].
Кроме медикаментозной терапии, как и при других заболеваниях легких, при развитии гипоксемии используется терапия кислородом. При развитии легочной гипертензии, кроме кислородотерапии, возможно использование вазодилататоров. Развитие инфекций трахеобронхиального дерева требуют использования антибактериальных и противогрибковых препаратов. Всем больным ИЛФ рекомендована регулярная вакцинация противогриппозными и антипневмококковыми вакцинами.
Другие идиопатические интерстициальные пневмонии (не-ИЛФ)
Неспецифическая интерстициальная пневмония (НИП) наряду с ИЛФ является одной из наиболее часто встречаемых форм ИИП. НИП может быть идиопатической, именно эта форма входит в группу ИИП. Однако морфологическая картина, соответствующая паттерну НИП, бывает и при поражении легких у больных с СЗСТ, гиперчувствительном пневмоните, радиационном пневмоните и т. д.
Клинические, лабораторные и функциональные показатели при НИП неспецифичны. Рентгенография грудной клетки чаще всего выявляет двусторонние изменения по типу «матового стекла» и ретикулярные изменения в нижних отделах легких.
Прогноз больных НИП более благоприятный, чем при ИЛФ. Клиническое течение и выживаемость больных зависят от выраженности легочного фиброза. Десятилетняя выживаемость при НИП составляет около 35%. Спонтанные случаи выздоровления без лечения при НИП неизвестны, терапия ГКС без или с добавлением цитостатиков приводит к улучшению или стабилизации приблизительно у 75% больных [9].
Криптогенная организующаяся пневмония
Синонимами криптогенной организующейся пневмонии (КОП) являются термины «облитерирующий бронхиолит с организующейся пневмонией» и «пролиферативный бронхиолит». КОП имеет четкие клинико-морфологические отличия от «изолированного» облитерирующего бронхиолита: наряду с поражением бронхиол наблюдается вовлечение в воспалительный процесс альвеол с наличием в их просвете организованного экссудата. КОП в большинстве случаев является идиопатическим, т. е. причина остается неустановленной. Среди установленных причин наибольше значение имеют СЗСТ (ревматоидный артрит и др.), осложнения лекарственной терапии (амиодарон, препараты золота и др.).
Заболевание чаще всего развивается у людей в возрасте 50–60 лет, мужчины и женщины болеют одинаково часто. КОП характеризуется острым или подострым течением, клиническая картина часто напоминает бактериальную пневмонию. Средняя продолжительность симптомов до момента постановки диагноза составляет 2–6 мес. Рутинные лабораторные тесты выявляют лейкоцитоз периферической крови (50%), повышение СОЭ и C-реактивного белка (70–80%).
Типичным рентгенологическим признаком КОП является наличие пятнистых, двусторонних (реже односторонних) плотных очагов консолидации субплевральной локализации. При КОП описана миграция легочных инфильтратов, чаще всего от нижних к верхним отделам. Дифференциальный диагноз КОП, кроме бактериальной пневмонии, проводят с хронической эозинофильной пневмонией, бронхоальвеолярным раком и лимфомой легких.
Спонтанное улучшение при КОП описано, но бывает редко. Терапия выбора при КОП — пероральные ГКС. Клиническое улучшение наступает уже через 1–3 суток от начала приема первой дозы, рентгенологические изменения обычно исчезают через несколько недель, общая длительность терапии ГКС составляет от 6 до 12 мес. При снижении дозы ГКС рецидивы заболевания возникают довольно часто, в такой ситуации вновь увеличивают дозу стероидов. Прогноз при КОП обычно благоприятный, большинство больных полностью излечивается при приеме ГКС. Однако в редких случаях наблюдается плохой ответ на стероиды и неуклонно прогрессирующее течение КОП. У таких больных рекомендовано использование цитостатиков [10].
Десквамативная интерстициальная пневмония
Десквамативная интерстициальная пневмония (ДИП) является довольно редким заболеванием из группы ИИП. Среди всех больных ДИП более 90% являлись курильщиками. Кроме того, описаны редкие случаи ДИП, ассоциированной с другими состояниями — СЗСТ, реакциями на лекарственные препараты, экспозицией к факторам внешней среды.
Клиническая картина заболевания типична для ИИП. Лабораторные, функциональные и рентгенологические показатели при ДИП не дают дополнительной информации.
При наличии сомнительной картины для исключения более агрессивных форм ИЗЛ рекомендовано проведение биопсии легких.
Отказ от курения является первым шагом лечения ДИП, так как показано, что данное мероприятие часто приводит к обратному развитию заболевания. Для большинства больных ДИП основным лечением является терапия преднизолоном в дозе 40–60 мг/сут. Начальная доза преднизолона обычно назначается на период 1–2 месяца, а затем дозу препарата постепенно снижают на протяжении 6–9 мес. На фоне терапии ГКС клиническое улучшение или стабилизация течения заболевания наблюдается приблизительно у двух третей больных ДИП. Значение цитостатиков при данной форме ИИП пока не ясно. 5- и 10-летняя выживаемость при ДИП составляет 95,2 и 69,6% соответственно [9].
Респираторный бронхиолит, ассоциированный с интерстициальным заболеванием легких
Респираторный бронхиолит, ассоциированный с интерстициальным заболеванием легких (РБ-ИЗЛ), — заболевание из группы ИИП, при котором респираторный бронхиолит сочетается с поражением альвеол и легочного интерстиция.
Данное заболевание встречается у курильщиков со стажем курения более 30 пачек/лет. Средний возраст больных колеблется от 30 до 40 лет. Клиническая картина и данные лабораторно-инструментального обследования типичны для ИИЛ.
Часто прекращение курения приводит к полному разрешению заболевания, в ряде случаев могут потребоваться небольшие дозы ГКС. Прогноз при РБ-ИЗЛ более благоприятный, чем при ИЛФ, но все-таки данное заболевание в ряде случаев может иметь неуклонно прогрессирующее течение и стать причиной смерти больных [11].
Лимфоцитарная интерстициальная пневмония
Лимфоцитарная интерстициальная пневмония (ЛИП) является одним из наиболее редко встречающихся заболеваний из группы ИИП. Как следует из названия, в основе заболевания лежит распространенная гомогенная лимфоцитарная инфильтрация легочного интерстиция. Морфологический диагноз ЛИП очень сложен, так как сходную гистологическую картину имеют некоторые заболевания, ассоциированные с массивной лимфоцитарной инфильтрацией ткани легких: псевдолимфома, первичная лимфома, лимфоматозный гранулематоз и др.
ЛИП встречается чаще всего у женщин, обычно в возрасте 40–60 лет. Большинство больных ЛИП — некурящие. Начало заболевания чаще всего незаметное, постепенное. Рентгенологическая картина ЛИП неспецифична.
Для постановки диагноза ЛИП во всех случаях требуется проведение открытой биопсии легких. Основу терапии ЛИП составляют ГКС. Дозы и длительность терапии приблизительно такие же, как и при других клеточных формах ИИП, например ДИП. На фоне противовоспалительной терапии улучшение или стабилизация заболевания отмечается у большинства больных (около 80%), хотя у небольшой группы их наблюдается медленное, но неуклонное прогрессирование заболевания. Кроме ГКС, у больных ЛИП применялись попытки терапии азатиоприном, циклофосфамидом, метотрексатом и циклоспорином [12].
Острая интерстициальная пневмония
Первые упоминания ОИП относятся к 1935 г., когда Hamman и Rich описали четырех больных с быстропрогрессирующей дыхательной недостаточностью, приведшей к смерти пациентов в течение 6 месяцев от начала болезни. На аутопсии был обнаружен выраженный распространенный фиброз легких [13]. Длительное время синдромом Хаммена–Рича назывались и заболевания с хроническим течением (в первую очередь ИЛФ), однако в настоящее время к синдрому Хаммена–Рича можно отнести только ОИП [14].
В современных руководствах ОИП рассматривается как заболевание, характеризующееся прогрессирующей дыхательной недостаточностью, приводящей в большинстве случаев к летальному исходу. Клиническая картина напоминает острый респираторный дистресс-синдром (ОРДС), однако при ОИП неизвестна причина заболевания и отсутствует вовлечение в процесс других систем организма (полиорганная недостаточность). В настоящее время в мировой литературе описано около 150 случаев ОИП, что связано не столько с редкостью заболевания, сколько со сложностью его диагностики [15].
Для ОИП характерно очень быстрое нарастание симптомов заболевания. Период от появления первых симптомов до обращения за медицинской помощью у большинства больных составляет не более 3 недель и очень редко превышает 2 месяца. Заболевание может развиться в любом возрасте и встречается одинаково часто у мужчин и женщин. Наиболее частыми симптомами ОИП являются непродуктивный кашель и диспноэ, лихорадка, миалгии, головная боль, слабость. При осмотре обращает на себя внимание тахипноэ, тахикардия, цианоз. При аускультации выслушивают крепитацию, реже — сухие свистящие хрипы.
Функциональные тесты неспецифичны и выявляют картину, характерную для других ИИП, однако полноценное функциональное исследование удается провести далеко не всегда. Характерным признаком ОИП является выраженная гипоксемия, часто рефрактерная к кислородотерапии, поэтому большинство больных, описанных в литературе, требовали проведения механической вентиляции легких.
Рентгенологическая картина при ОИП выявляет двусторонние пятнистые ретикулонодулярные тени, распространяющиеся практически на все легочные поля, за исключением реберно-диафрагмальных синусов, и плотные инфильтраты (консолидация). Типичными находками компьютерной томографии легких являются участки пониженной прозрачности паренхимы по типу «матового стекла», дилатация бронхов и нарушение легочной архитектоники. Изменения по типу «матового стекла» чаще всего имеют пятнистое распространение («географическая карта»).
Для морфологической верификации диагноза возможно проведение открытой или торакоскопической биопсии легких. Однако, к сожалению, из-за крайней тяжести больных с ОИП проведение данной диагностической процедуры чаще всего бывает невозможно. Все описанные в литературе морфологические изменения ОИП основаны на данных аутопсии или открытой биопсии легких, выполненной во время проведения больным ИВЛ.
Заболевание характеризуется фульминантным течением, прогноз плохой, летальность больных ОИП крайне высока и составляет, в среднем, 70% [16]. Дифференциальный диагноз ОИП чаще всего проводится с двусторонней бактериальной пневмонией или ОРДС. При ОРДС, как правило, известна причина (сепсис, травма, шок и т. д.); кроме того, ОРДС чаще всего бывает одной из составных частей полиорганной недостаточности.
Эффективной терапии ОИП в настоящее время не существует. Обязательными компонентами терапии ОИП являются кислородотерапия и респираторная поддержка.
Литература
М. В. Вершинина, кандидат медицинских наук, доцент
ГБОУ ВПО ОмГМА Минздравсоцразвития России, Омск



