Хромосомные нарушения
Хромосомные нарушения — это клинические синдромокомплексы, в основе которых лежат нарушения числа или структуры хромосом, то есть избыток или нехватка генетического материала, локализованного в той или иной хромосоме.
В норме у человека число хромосом равно 46, из которых 23 ребенок получает от матери и 23 аналогичные хромосомы от отца. В этом наборе гентического материала есть 2 особые хромосомы, которые были названы «половыми». Они определяют пол ребенка и ряд других важных признаков.
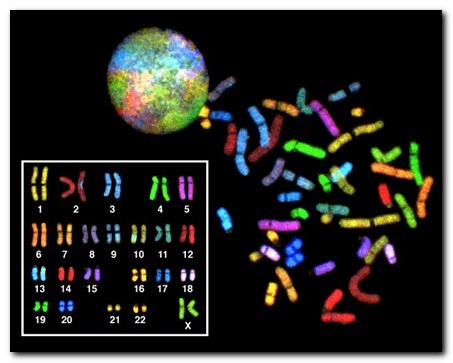
Таким образом, изменения числа хромосом (больше или меньше 46), а также изменение структуры хромосом (например, выпадение или удвоение даже небольшого кусочка хромосомы) получили название «хромосомные мутации».
Наиболее часто из них встречаются изменения модального числа хромосом — это отсутствие в хромосомном наборе какой-либо хромосомы (моносомия) или появление добавочной хромосомы (трисомия, тетрасомия и т.д.).
Число возможных изменений структуры хромосомы неисчислимое множество. К примеру, транслокации (обмен сегментами между разными хромосомами), делеции (выпадение участка хромосомы), дупликации (удвоение части хромосомы), инверсии (переворот сегмента хромосомы на 180 градусов) и т.д.
Хромосомные мутации, возникшие в половых клетках (сперматозоидах или яйцеклетках) или на первых этапах деления клеток зародыша, как правило, передаются большинству клеток развивающегося организма, вызывая множественные аномалии развития, а многие хромосомные изменения плода могут стать причиной спонтанных абортов и выкидышей, что важно учитывать в семьях, воспитывающих детей с задержками развития.
К факторам риска, способствующим их возникновению, относят ионизирующую радиацию, инфекции и интоксикации матери, эндокринные нарушения, психические травмы, воздействие ряда лекарственных препаратов и некоторых физиотерапевтических методов лечения.
Наиболее точно установлено, что причиной появления ребенка с хромосомными мутациями является не молодой возраст матерей (свыше 40 лет).
В последнее время очень большое значение придается факту скрытого носительства хромосомных нарушений у родителей родившегося ребенка (сбалансированные транслокации, мозаицизм). Изучение данного вопроса позволяет предотвратить риск повторного рождения ребенка с аналогичной формой заболевания.
Различают хромосомные синдромы, обусловленные изменением половых хромосом, и синдромы, вызванные аномалиями аутосом (любой из 44 неполовых хромосом).
Основными клиническими проявлениями аутосомных аномалий являются признаки психического и физического недоразвития, дисплазии (неправильное развитие), врожденные пороки развития (аномалии) и умственная отсталость различной степени тяжести. К врожденным порокам можно отнести: аномалии развития сердца, удвоение почки, расщелина неба, особенности строения кистей и стоп и многие другие. При заболеваниях, обусловленных нарушениями в системе половых хромосом, как правило, более характерны недоразвитие половых желез и аномалии развития вторичных половых признаков, также с симптомами задержки психо-речевого развития.
Различные хромосомные синдромы встречаются с разной частотой. По сводным данным многих исследований, распространенность наиболее частых из них среди новорожденных следующая:
трисомия по 21 хромосоме (синдром Дауна) 1:500
XXX (трисомия-Х) 1:1000 (девочек)
ХYY (синдром дубль-Y) 1:1000 (мальчиков)
ХХY (синдром Клайнфелтера) 1:1400 (мальчиков)
Х0 (синдром Шерешевского-Тернера) 1:3300 (девочек)
46,5р del (синдром «кошачьего крика») 1:4000
трисомия по 18 хромосоме (синдром Эдвардса) 1: 6800
трисомия по 13 хромосоме (синдром Патау) 1:7600

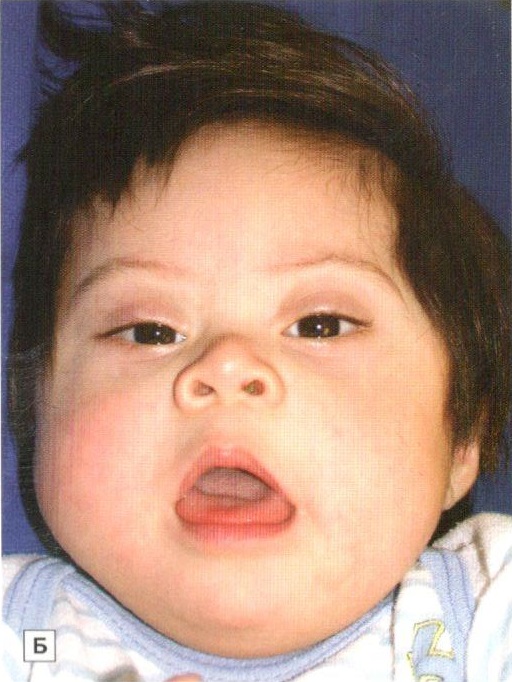
Установлено, что для синдрома Дауна характерно уменьшение размеров и веса головного мозга, а также аномалии развития мозга и мозговых сосудов. Отмечаются также структурные изменения в железах внутренней секреции, печени и сердце. Клиническая картина синдрома Дауна характеризуется проявлениями симптомов умственной отсталости. Характерен также и внешний вид таких больных: косо расположенные глазные щели, широкая уплощенная переносица, дополнительная кожная складка у внутреннего угла глаз, высокое стояние твердого неба (признаки эмбриональной задержки в развитии лицевого скелета), полуоткрытый рот, увеличенный высунутый язык с выраженными сосочками и глубокими бороздами (признаки дисфункции щитовидной железы), выпадение волос (дисфункция надпочечников), низкий рост, короткая шея, укороченные кисти и стопы, искривление мизинца, на ладонях имеется поперечная складка, на стопах увеличен промежуток между 1 и 2 пальцами, выражены внешние проявления гипогенитализма.
В клинической картине заболевания доминируют симптомы неврологической патологии, диффузная мышечная гипотония (снижение мышечного тонуса), благодаря чему больные гибки и иногда могут складываться как «перочинный ножик», расстройства координации движений, косоглазие, выраженные вегетососудистые нарушения.
Особенностью психического дефекта является относительная сохранность эмоциональной сферы по сравнению с тяжестью интеллектуального недоразвития. Так, больные ласковы, добродушны, послушны. Характерной особенностью таких детей является повышенная внушаемость, что является положительным фактором при проведении коррекционной работы и отрицательным при их развитии.
Уровень социального развития больных с синдромом Дауна зависит от степени и формы заболевания. Так, дети с более легкими формами умственной отсталости, хотя и медленно, но развиваютя, приобретая определенные навыки, знания, осваивая программу нескольких классов вспомогательной школы. Однако, как правило, большинство из них не достигают удовлетворительного уровня социальной адаптации и нуждаются в постоянной опеке. Им может быть оформлена инвалидность детства с момента точной диагностики заболевания. Особенностью возрастной динамики синдрома Дауна является позднее половое созревание и раннее появление признаков инволюции (25—30 лет). Мужчины с синдромом Дауна бесплодны, женщины могут давать потомство, половина которого также страдает синдромом Дауна.
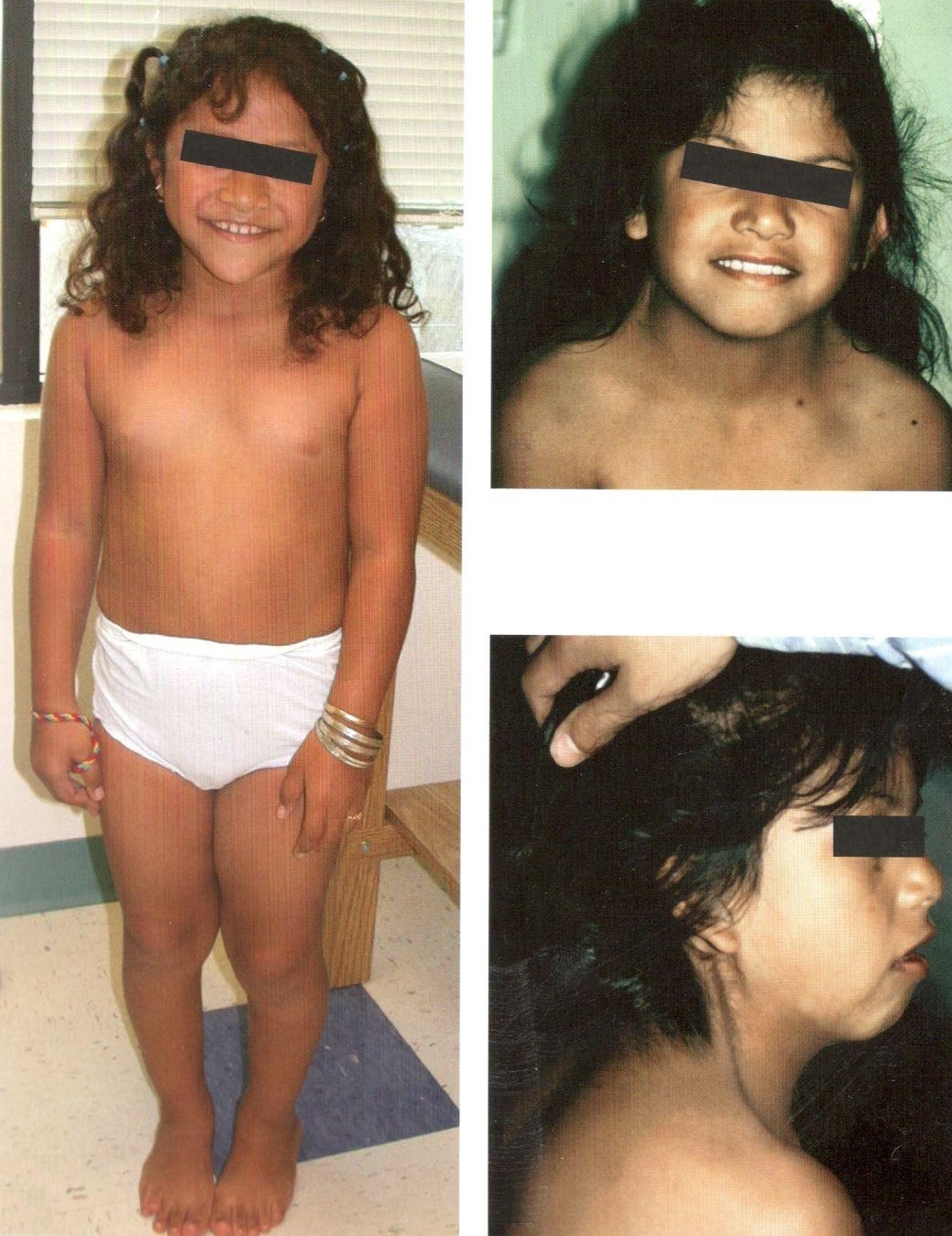
Впервые заболевание описано отечественным эндокринологом Н.А. Шерешевским (1925), а более подробно — американским эндокринологом Н. Тернером (N.H. Terner) л 1938 г. В основе заболевания лежит отсутствие одной хромосомы (половой Х-хромосомы) (45 вместо 46).
Клиническая картина синдрома характеризуется разной степенью умственной отсталости и ЗПРР, низким конечным ростом (135—145 см), замедлением полового развития, недоразвитием половых желез, аменореей, бесплодием и отсутствием грудных желез. Диспластические расстройства проявляются в виде короткой шеи и особых кожных складок, идущих от затылка к надплечью, укорочением 4 пальцев на руках и искривлением мизинцев, выраженной деформацией ушных раковин, наличием множественных пигментных родинок. Преимущественно данным синдромом страдают лица женского пола.
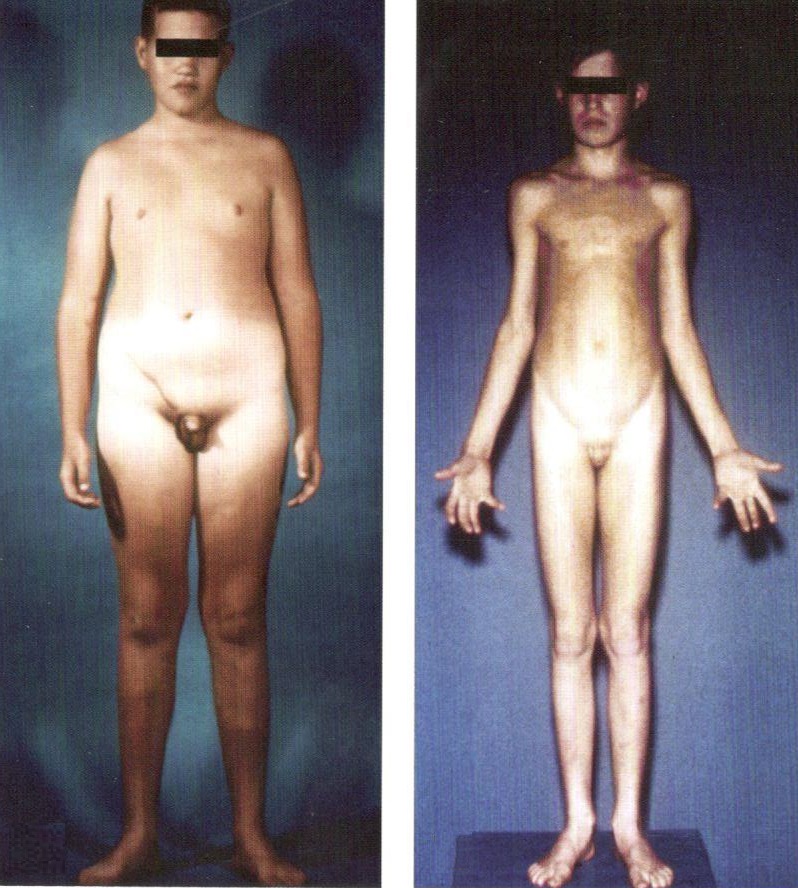
Клинические проявления синдрома Клайнфельтера варьируют от внешне нормального и интеллектуального развития до выраженного евнухоидизма и умеренной умственной отсталости. Однако в ряде случаев уже в раннем возрасте у больных отмечаются характерные своеобразные симптомы физического развития: низкий и узкий лоб, густые и жесткие волосы, высокое стояние таза, короткая, плоская и узкая грудная клетка, недоразвитие половых органов. Более отчетливо вышеперечисленные симптомы начинают обнаруживаться в подростковом, пубертатном возрасте. Характерен внешний вид взрослого больного с синдромом Клайнфельтера: высокий рост, астеническое сложение, узкие плечи, широкий таз, удлиненные конечности, слаборазвитая мускулатура, скудная растительность на лице и в подмышечных впадинах, ожирение и оволосение по женскому типу, сутулость, выраженные евнухоидные пропорции и гинекомастия (набухание грудных желез). Постоянными признаками синдрома Клайнфельтера являются недоразвитие половых органов и бесплодие.
Степень интеллектуального недоразвития у больных выражена тем глубже, чем больше дополнительных половых хромосом обнаруживается в кариотипе (46 или 49). Так, умеренная умственная отсталость зачастую приближается к психическому инфантилизму, что клинически проявляется недостаточностью внимания, восприятия, памяти, абстрактного мышления, чрезмерной внушаемостью, подражательностью, подчиняемостью, несамостоятельностью, чрезмерной привязанностью к близким, нередко с элементом назойливости. Глубокая незрелость эмоционально-волевой сферы проявляется в виде повышенного настроения, с эйфорическим оттенком, склонностью к эксплозивным аффективным вспышкам, неспособностью к длительному волевому усилию и напряженной деятельности. У больных, как правило, отсутствуют чувство долга и ответственности. При легких формах заболевания больные осознают свою неполноценность, что приводит к внутреннему конфликту и возникновению у них невротических реакций. Данным синдромом страдают лица мужского пола.
Синдром ломкой Х-хромосомы (Fragile X syndrome, FraХ). Начиная с 1980 года большое значение придают синдрому ломкой Х-хромосомы (Хq27.3) – именно с ним связывают развитие более чем 50 наследственных расстройств, включая ранний детский аутизм и 30% случаев умственной отсталости у мальчиков. Хрупкий участок Х-хромосомы впервые обнаружил Labs (1969).
Полная мутация в Х-хромосоме возникает только у женщин, и происходит это в процессе гаметогенеза, поэтому почти всегда страдают мальчики, получившие единственную Х-хромосому от матери. У девочек, получивших вторую Х-хромосому от отца, также могут быть нарушения развития, но они менее выражены, а тяжелые патологии встречаются много реже, чем у мальчиков. В отдельных случаях девочки могут получить обе ломкие хромосомы от матери, в этом случае частота и тяжесть патологии будет одинаковой с мальчиками.
Клиническую триаду синдрома ломкой Х-хромосомы образуют:
1) умеренная до степени тяжелой умственная отсталость. Лишь 30% лиц мужского пола имеют интеллект, стремящийся к нижней границе нормы, а среди женщин – носительниц такой хромосомной патологии примерно у 30% обнаруживаются признаки умственного недоразвития;
2) характерные особенности строения лица и черепа: выдающийся вперед высокий лоб, прогнатизм и удлиненные уши;
3) мальчики имеют увеличенные в размерах тестикулы (макроорхидизм).
Наблюдаются, кроме того, эпилептические припадки, синдром гиперактивности с дефицитом внимания, у более чем половины мальчиков аутизм и подобные аутизму расстройства, различные нарушения развития речи, персеверации, эхолалия, другие отклонения.
Женщины, унаследовавшие ломкую Х-хромосому с полной мутацией от своих матерей, могут быть склонны к развитию атипической депрессии, а также шизофреноподобного заболевания.
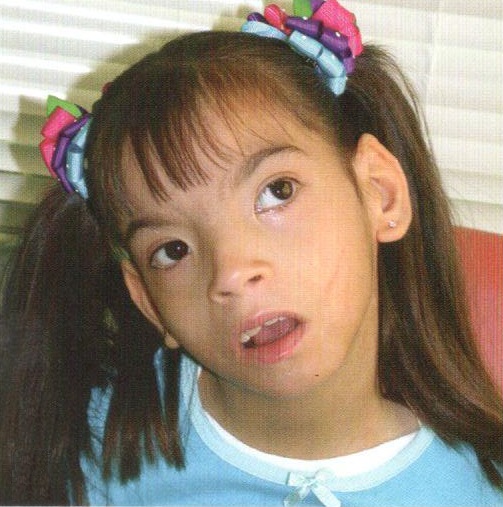
Синдром Вольфа—Хиршхорна.
В основе синдрома лежит изменение длины хромосомы из четвертой пары. Основные признаки заболевания у новорожденных: большое туловище, клювовидный нос и выступающее надпереносье, деформированные ушные раковины со складками, пучеглазие и колобома радужной оболочки (ее частичное отсутствие), общее недоразвитие во время беременности. Отмечается наличие четырех сгибательных складок на пальцах верхних конечностей.
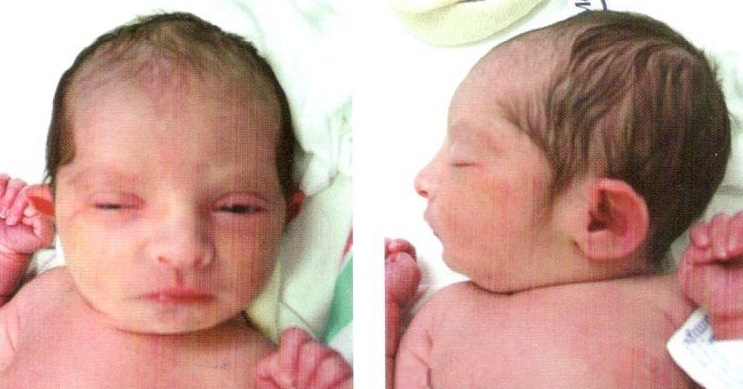
Клиническая картина характеризуется микроцефалией, расщелиной лица, двусторонним расщеплением верхней губы, полным расщеплением неба, маленькими глазными яблоками либо полным их отсутствием, короткой шеей, маленькими деформированными низко расположенными ушами, полидактилией, дистрофическими изменениями ногтей и костного скелета. Отмечаются также пороки развития сердца, желудка, кишечника и других органов.
Синдром трисомии-Х впервые описан в 1959 г. Частота данной патологии составляет среди новорожденных 0,1%, а среди умственно отсталых — 0,6%. Большинство лиц женского пола с трисомией-Х выявляется среди больных психиатрических лечебниц. Клиническая картина характеризуется аномалиями развития скелета, внутренних органов, различными психическими проявлениями и интеллектуальной недостаточностью. Среди полиморфизма признаков трисомии-Х наиболее характерными являются: низкий рост, аномалии ушей, прикуса, высокое стояние твердого неба, короткие пальцы, искривленный мизинец, широкий промежуток между 1 и 2 пальцами на стопах, синдактилия, недоразвитие половых функций.
Умственная отсталость проявляется в виде легкой или умеренной степени. Характерны эмоциональные расстройства (вспыльчивость, агрессивность, неустойчивость настроения и немотивированные поступки). Девочки с синдромом трисомии-Х с трудом, но в большинстве случаев (легкая степень умственной отсталости) обучаются в массовых школах.
К хромосомным синдромам, помимо вышеописанных, относится большая группа так называемых семейных форм умственной отсталости, когда совершенно точно доказано наличие данной патологии у близких родственников.
Синдром Аперта (акроцефалосиндактилия) — наследственное заболевание, характеризующееся умеренной или тяжелой умственной отсталостью, экзофтальмом, деформацией зубов и синдактилиями. Синдром описан французским педиатром Апертом (Е. Apert) в 1906 г.
Синдром Крузона — наследственное заболевание, характеризующееся умеренной или тяжелой умственной отсталостью, преждевременным срастанием швов черепа, уменьшением мозгового вещества, экзофтальмом, вторичной атрофией зрительных нервов, прямоугольным расположением большого пальца к кисти. Впервые синдром описан французским врачом Крузоном (О. Crouson) в 1912 г.
Синдром Берьесона—Форсмана—Лемана — синдром характеризующийся умственной отсталостью в сочетании с избыточным весом. Впервые описан американскими врачами Берьесоном (М. Berjeson) Форсманом (Н. Foreman) и Леманом (О. Lehman) в 1963 г. Клиническая картина заболевания проявляется выраженным ожирением и прогрессирующей умственной отсталостью. Ожирение носит не равномерный характер. Жир откладывается преимущественно на бедрах, груди и лице, что придает своеобразный вид такому больному (бочкообразная карликовая фигура с заплывшим лицом, большими ушами и узкими разрезами глаз). У больных часто отмечаются эпилептические припадки. Умственная отсталость колеблется от умеренной до тяжелой степени. Данная патология встречается только у лиц мужского пола, но носителями патологического гена являются женщины.
Синдром Прадера—Вилли — наследственное заболевание, характеризующееся глубокой умственной отсталостью, низким ростом, гипогенитализмом, ожирением, резко выраженной мышечной гипотонией.
Синдром Книппеля—Фейля (синдром короткой шеи) — наследственное семейное заболевание, обусловленное врожденными аномалиями развития скелета и внутренних органов в сочетании с тяжелой степенью умственной отсталости. Клиника синдрома подробно описана французскими врачами Клиппелем Фейлем в 1912 г.
Аномалия развития характеризуется следующими проявлениями: короткой шеей как результат количественного уменьшения шейных позвонков, ограничением подвижности головы, расщеплением твердого неба, бочкообразной грудной клеткой, врожденными пороками сердца, добавочными долями или отсутствием отдельных долей легких, синдактилиями (сращение пальцев конечностей), глухотой вследствие заращения наружных слуховых проходов, сужением анального отверстия и многими другими симптомами. Интеллектуальная недостаточность является результатом тяжелой умственной отсталости
Лечение ЗПРР при хромосомных заболеваниях.
Исследования последних десятилетий выявили, что у большинства детей с речевыми и поведенческими проблемами в различной степени нарушены функции мозжечка и базальных ганглиев. Именно функционирование мозжечка определяет успешность ребенка в обучении. С этой целью применяется уникальная разработка Центра авиакосмической медицины — подошвенный имитатор опорной нагрузки «Корвит», применяемый для нейрофизиологической регуляции стато-кинетической функции ЦНС. В основе терапевтического воздействия аппарата «Корвит» лежит процесс активации опорной афферентации, отвечающей за нормализацию процессов возбуждения и торможения в центральной нервной системе, что приводит к уменьшению спастичности мышц, развитию и закреплению функциональных связей в головном мозге, способствующих восстановлению координации движений, и, опосредованно, улучшению речи и мышления.
Обязательным звеном в лечебном комплексе у детей с наличием речевых расстройств является занятия с клиническим психологом, а также логопедическая коррекция, которая включает диагностику степени нарушений, ежедневные занятия, направленные на улучшение речевой функции и логопедический массаж для коррекции различных видов дизартрии и дисфагии.
На фоне сочетания проведения биофизической активации со вспомогательными методиками лечения наблюдаются положительные изменения, которые могут быть видны уже через несколько процедур, но максимальный эффект развивается через полтора-три месяца после курса. Как правило, для закрепления полученных результатов и дальнейшего развития двигательных и когнитивных навыков специалистами центра рекомендуется повторный курс лечения через 5-6 месяцев.
Микроделеционные синдромы
Организм человека состоит из огромного количества клеток. Они в свою очередь объединяются в более крупные единицы – ткани и органы. Из последних строятся системы органов, а и из них и получается организм. И на всех уровнях постоянно идут различные взаимодействия и сложные биохимические реакции. Чтобы все это гармонично развивалось из оплодотворенной яйцеклетки и слаженно функционировало, требуется специальная схема работы для всех уровней. Ее роль в организме выполняет организованная генетическая информация.
Именно на наследственный материал возлагается задача по сохранению данных об общем облике человека, особенностях строения отдельных органов, принципах регуляции при помощи гормонов, и даже того, каким образом должны собираться белки. Все данные записываются в виде последовательности структурных единиц – нуклеотидов. Они подобно буквам алфавита формируют группы-слова, выполняющие определенные функции. Каждая группа называется геном. Все вместе они формируют молекулу ДНК. Если посмотреть на уровень выше, то можно заметить, что весь наследственный материал в виде цепочек дезоксирибонуклеиновой кислоты сконцентрирован в особых образованиях, которые называются хромосомами.
Их у человека в норме насчитывается 23 пары. В каждой паре информация хранится в двух копиях. Это необходимо для того, чтобы при передаче генетической информации каждая дочерняя клетка получила свою копию. Одна пара хромосом отличается от остальных и отвечает за вопросы определения пола. Если в ней имеются две одинаковые большие хромосомы (ХХ вариант), то организм относится к женскому полу. Если же имеется одна большая и одна малая хромосома, то речь идет о мужском поле (XY вариант). Эти две отличающиеся от прочих хромосомы называются половыми. Оставшиеся 22 пары встречаются во всех организмах независимо от пола. Их называют аутосомами.
Аномалии генетического материала

Наследственный материал состоит из огромного количества нуклеотидов, формирующих гены. При этом в каждом гене последовательность нуклеотидов строго определена, поскольку должна кодировать определенный белок. Кроме того, сами гены при формировании хромосом также выстраиваются в фиксированном порядке. Благодаря сохранению этого порядка организм может функционировать, а ученые – быстро и точно указывать друг другу, про какой ген идет речь.
В идеальном случае система работает без малейших сбоев, а генетическая информация всегда передается в неизменном виде. Однако на практике большое число структурных единиц и постоянное воздействие различных факторов (например, ионизирующего излучения) приводит к тому, что время от времени возникают различные аномалии. В частности, отдельные участки последовательности ДНК могут быть скопированы на новое место. В таком случае говорят о дупликации. Если же вместо создания новой копии была перемещена часть исходной цепочки, то модификация называется транслокацией. Кроме того, иногда часть последовательности просто теряется, удаляется из генетического материала. В таком случае изменение называется делецией.
Поскольку взаимодействия в организме оттачивались в течение многих тысячелетий эволюционного развития, получилась очень слаженная система. И аномалии, даже самые небольшие, могут вызвать нарушение баланса. В таком случае в организме развивается то или иное нарушение. Если при этом причина находится на уровне генов, то говорят о генных болезнях. Если была утрачена или наоборот получена лишняя копия хромосомы, то такие нарушения называются хромосомными заболеваниями.
Что такое микроделеционный синдром?
Самые незначительные изменения (они же мутации) называются точечными. Их появление влияет на считанные единицы генов. В некоторых случаях нарушение относится вообще к одному единственному гену. Однако если он обеспечивал выработку важного белка, последствия для всего организма могут быть очень серьезными. Подобные патологические изменения относятся к группе микроделеционных синдромов.
Каждое такое заболевание обусловлено небольшим изменением генетического материала, которое происходит в строго определенном месте. Точный механизм возникновения подобных нарушений на сегодняшний день не установлен, что не мешает ученым заниматься исследованием их воздействия на организм.
Так, было выяснено, что развитие синдрома в таком случае может происходить несколькими различными вариантами. В частности, ряд заболеваний характеризуется участием онкогенов. В других случаях на воздействие непосредственно самой делеции накладывается эффект хромосомного импринтинга и возможные однородительские дисомии.
Частота возникновения большей части микроделеционных синдромов крайне невелика: порядка 1 случая на 50-100 тысяч новорожденных. Набор клинических признаков обычно выражается отчетливо. Для того чтобы поставить диагноз, бывает достаточно лишь совокупности симптомов. Однако при таком подходе невозможно точно прогнозировать здоровье потомков, поэтому зачастую наряду с проверкой обычных признаков производится молекулярно-генетическая диагностика пробанда и его родственников (обычно родители, в некоторых случаях также требуется анализ генотипа братьев, сестер, теть, дядь и так далее).
Патологические проявления сильно отличаются. В частности, их проявление определяется тем, насколько большой участок генетического материала был утрачен в результате делеции. Кроме того, в ряде случаев играет роль то, от кого из родителей была получена мутация (влияние хромосомного импринтинга). Хорошей иллюстрацией последней ситуации является пара синдромов Прадера-Вилли и Ангельмана. Они оба обусловлены наличием делеции в 15 хромосоме. Однако из-за различного механизма действия при передаче от разных родителей клиническая картина этих заболеваний значительно отличается.
Позитивное влияние некоторых делеций на жизнеспособность
Небольшие изменения делеционного характера могут существенно повлиять на выживание организма. К примеру, утрата гена, кодирующего белок CCR5-δ32, становится причиной невосприимчивости к вирусу иммунодефицита человека. Ученые предполагают, что эта мутация впервые появилась около 2,5 тысячелетий назад и с течением времени распространилась по территории Европы.
Имеющееся на сегодняшний день распределение отличается неравномерностью. Согласно статистическим данным, около 10% жителей европейских стран устойчиво к ВИЧ. Вместе с тем в скандинавских государствах этот показатель достигает 14-15 процентов. Русские и финны демонстрируют 16-процентный уровень устойчивости. В то же время для Сардинии частота равняется скромным 4 процентам.
Ряд ученых выдвинул гипотезу, что подобное распространение определяется прошедшими в средневековый период эпидемиями бубонной чумы. Вероятно, мутация в гене вызывает повышенную сопротивляемость этому заболеванию. Поэтому на территории тех стран, где прошлась «черная смерть», выжило больше людей с этим генотипом.
Влияние делеций на способность к оплодотворению
Делеции, происходящие в обычных хромосомах (аутосомах) могут в некоторых случаях быть компенсированы нормальной копией гена. Однако когда речь заходит о половых хромосомах, особенно об Y-хромосоме, ситуация меняется.
Прежде всего необходимо отметить, что локализованные на ней гены не имеют второго экземпляра. При нормальном количестве хромосом в наборе Y-хромосома оказывается крайне уязвимой. В сочетании с малым количеством генов это приводит к серьезным последствиям каждого изменения. Особый интерес представляют мутации, касающиеся AZF-локуса и SRY гена.
Аномалии гена SRY (Sex-determining Region Y)
Ген SRY, как следует из его названия, отвечает за крайне важную функцию. Именно его наличие в хромосомном наборе запускает процесс формирования организма по мужскому фенотипу и стимулирует развитие соответствующих половых органов.
Наличие даже небольшой делеции в этом гене нарушает механизм дифференцировки пола. В результате при нормальном кариотипе 46XY зародыш начинает развиваться как женский организм. По этой причине на ген SRY приходится наибольшее число мутаций, связанных с неразвитостью гонад. Кроме того, изменения этого гена вызывают инверсию пола.
Аномалии AZF-локуса
На Y-хромосоме также имеется особый участок, который контролирует процесс выработки сперматозоидов. Именно от этого зависит, насколько эффективным будет сперматогенез. Кроме того, состояние этого участка сказывается на свойствах сперматозоидов, таких как общее количество в эякуляте, способность двигаться, наличие структурных изменений и способность к оплодотворению. Только при наличии хорошо сформированных подвижных сперматозоидов мужской генетический материал может быть доставлен до яйцеклетки. Иными словами, о состоянии этого небольшого участка генетического кода зависит способность мужчины иметь детей.
При наличии в AZF-локусе аномалий процесс выработки сперматозоидов нарушается. В результате могут развиться азооспермия и олигозооспермия. При этих патологиях в эякуляте либо совсем не содержится сперматозоидов, либо их число сильно снижено.
Сам AZF локус делится на три части со специфическими задачами. Они именуются путем добавления индекса: AZFa, AZFb и AZFc. Возникшая делеция может удалять фрагмент отдельной части, либо ее целиком, либо захватывать сразу два региона. При полном удалении AZF развивается тяжелое поражение сперматогенеза. Частичные делеции могут проявляться по-разному. При этом на степень проявления патологии влияют размеры утраченного фрагмента и его расположение в локусе. Поэтому для прогностических целей крайне важно знать, в каком месте произошла делеция. Кроме того, эта информация может использоваться для правильного планирования семьи и проведения экстракорпорального оплодотворения.
Если при делеции был удален весь локус или любой из регионов с индексами a/b, то у мужчины не могут быть получены жизнеспособные сперматозоиды. Если делецию можно описать формулой AZFb/AZFb+, то развивается азооспермия из-за тяжелых нарушений процесса формирования сперматозоидов.
Делеции участка AZFc приводят к проявлению патологических симптомов различной степени тяжести. В том числе возможно развитие олигоспермии, которая в принципе допускает зачатие. В 50-70 процентах от общего числа подобных случаев возможно получение сперматозоидов для дальнейшего использования в методах искусственного оплодотворения. Частичная делеция региона AZFс может выражаться в форме различных нарушений от нормозооспермии до азооспермии.
Все делеции в AZF-локусе, вызывающие ту или иную патологическую ситуацию, являются причинами мужского бесплодия. Определение мутации возможно путем гистологического анализа семенной жидкости. При этом необходима остановка созревания сперматозоидов или обнаружение незрелых сперматозоидов. Для получения точных данных о делециях в AZF-локусе используется ПЦР 6 маркеров, которые относятся к отдельным участкам локуса.
Синдром Ангельмана

При синдроме Ангельмана развивается характерный набор патологических изменений. В частности, отмечается задержка психологического развития, сопровождающаяся проблемами со сном, частыми хаотическими движениям (больше руками), постоянными улыбками и смехом.
Патология развивается при отсутствии некоторых генов, расположенных на 15 хромосоме. При этом обязательным условием является передача мутантной копии гена от матери. Если поврежденная хромосома будет унаследована от отца, то разовьется синдром Прадера-Вилли. Кариотип обычно нормальный (46XX и 46XY для девочек и мальчиков соответственно). Различные независимые исследования указывают на связь болезни с геном UBE3A, который в норме обеспечивает выработку ферментного компонента в сложной системе деградации белков.
Частота появление синдрома составляет примерно 1 случай на 10-20 тысяч новорожденных (показатели отличаются у различных ученых).
Характерными особенностями больных с синдромом Ангельмана являются следующие признаки:
· проблемы с питанием, начинающиеся еще во время грудного вскармливания, поскольку дети плохо набирают вес (распространенность признака порядка 75 процентов);
· заторможенное развитие навыков общей моторики, то есть дети позже других начинают сидеть и ходить;
· для всех детей характерны нарушения речевого развития;
· больные обычно понимают больше, чем в состоянии выразить при помощи ограниченного словарного запаса;
· часто заболевание сопровождается дефицитом внимания и гиперактивностью;
· проблемы с обучением в обычной школе;
· у 80% заболевших развивается эпилепсия, сопровождающаяся заметными на электроэнцефалографии нарушениями; ученые полагают, что заболевание эпилепсией носит вторичный (симптоматический) характер.
· выполнение необычных движений, к которым относятся произвольные хаотические движения конечностями, мелкий тремор;
· возникновение приступов смеха при отсутствии видимых причин;
· характерная ходьба на негнущихся ногах, из-за которой возникло сравнение с марионетками;
· уменьшенная по сравнению со средними размерами голова, часто с уплощенным затылком;
· в некоторых случаях встречаются своеобразные запоминающиеся черты лица – широкий рот с редко расположенными зубами, выдвинутый вперед подбородок с выпущенным наружу языком;
· различные нарушения сна;
· примерно в 40 процентах случаев развивается косоглазие;
· порядка 10% больных также страдает от искривления позвоночника;
· высокие температуры воспринимаются с повышенной чувствительностью;
· наибольшего комфорта обычно достигают в воде (к примеру, в ванной)
Как правило, синдром определяется при помощи методов молекулярно-генетической диагностики по 15 хромосоме. Показанием к проведению тестирования для новорожденного является пониженный мышечный тонус (гипотонус), заметное отставание в развитии речи и мелкой моторики. Кроме того, на заболевание могут указывать мелкий тремор, порывистые беспорядочные движения, передвижение на негнущихся ногах.
Анализ может проводиться через флуоресцентную гибридизацию in situ, метилированием ДНК в области 15q11-q13. Также можно проверить мутации в импринтинговом центре и в гене UBE3A.
Поскольку заболевание обусловлено генетическим нарушением, адекватного и действенного способа лечения для него не имеется. Выполнение лечебных мероприятий, таких, как массаж для больных с гипотонусом, позволяет повысить качество жизни.
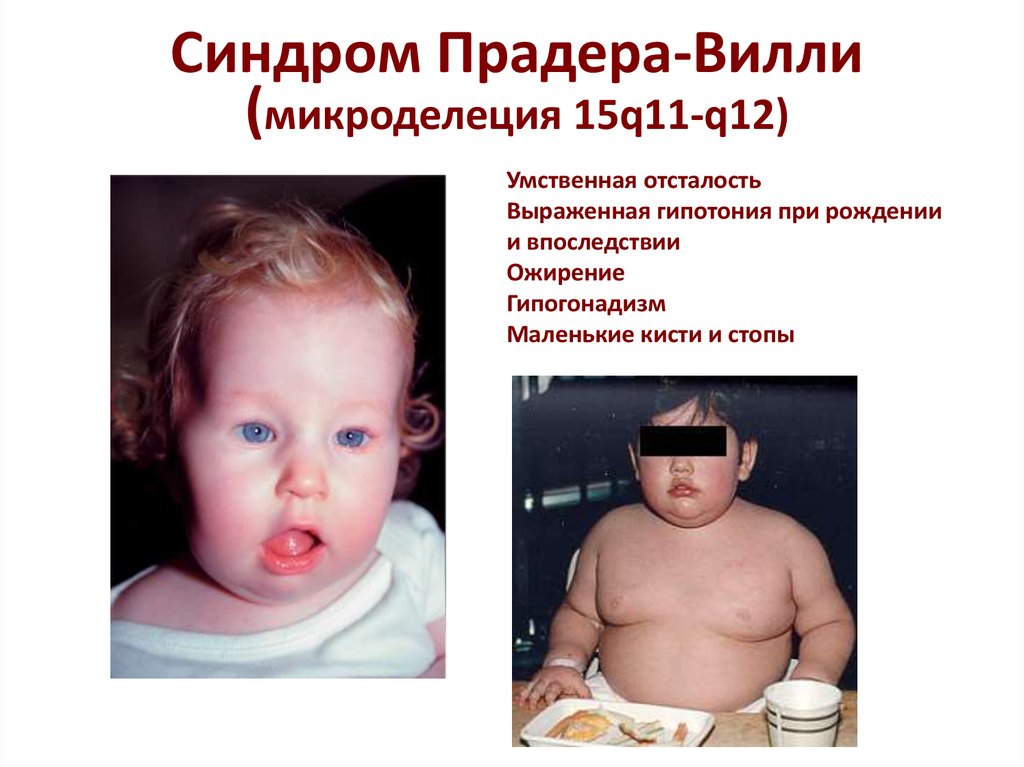
Синдром Прадера-Вилли
Это заболевание определяется той же самой генетической мутацией, что и для синдрома Ангельмана. Отличие состоит в том, что при этом нарушение наследственного материала получается со стороны отца. Кариотип соответствует нормальному (46XX или 46XY). По распространенности (1 случай на 12-15 тысяч новорожденных) примерно совпадает с распространенностью синдрома Ангельмана.
Характерными признаками синдрома Прадера-Вилли являются следующие симптомы:
· в пренатальный период малая подвижность плода;
· часто встречается неверное положение плода;
· возможна дисплазия тазобедреных суставов;
· к двум годам может проявиться склонность много есть (больше нормы), что приводит к ожирению;
· низкий мышечный тонус (гипотонус), сочетающийся с нарушенной координацией движений;
· стопы и кисти обычно маленькие, кроме того характерен невысокий рост;
· формирование косоглазия и сколиоза;
· отмечают повышенную сонливость;
· плотность костей находится на более низком уровне, чем у здоровых людей;
· слюна густая, обычно состояние зубов плохое;
· недостаточная функция половых желез, вызывающая в итоге бесплодие;
· позднее по сравнению со сверстниками половое созревание;
· больные позже учатся говорить, отстают в психическом развитии;
· внешние признаки включают выраженную переносицу, узкий и высокий лоб, миндалевидную форму глаз, узкие губы.
В большинстве случаев у человека с мутацией насчитывается от одного до пяти признаков заболевания.
Диагностика заболевания проводится путем молекулярно-генетического тестирования, на которое направляются дети с пониженным мышечным тонусом. Зачастую вместо верного диагноза определяется более распространенный «синдром Дауна». Опытный генетик, достаточно часто встречающийся с проявлениями синдрома Прадера-Вилли способен диагностировать его по комплексу внешних признаков.
Синдром лиссэнцефалии Миллера — Дикера
При синдроме лиссэенцефалии Миллера – Дикера причиной патологических изменений является делеция некоторых генов в локусе 17p13. При этом больше всего страдает центральная нервная система. Наряду с лиссенцефалией (сглаживание находящихся на поверхности мозга извилин из-за нарушения деятельности гена PAFAH1B1) отмечается сокращение числа кортикальных слоев. Если в норме их насчитывается 6 штук, то у больных можно обнаружить только 4. Сопутствующими признаками является заметное изменение форм лица. Кроме того, больные медленно растут. Попытки интеграции в общество осложняются множественными патологиями сердца, желудочно-кишечного тракта, почек. Если при заболевании происходит делеция гена 14-3-3 эпсилон, то синдром проявляется значительно тяжелее.
Аниридия
При аниридии нарушается нормальное строение глаза: в органе зрения отсутствует радужная оболочка. Кроме того, часто развиваются сопутствующие патологические изменения, такие как макулярная гипоплазия и гипоплазия зрительного нерва, изменения роговицы, катаракта. Острота зрения заметно падает, попытки коррекции не приносят существенных результатов. Развивается светобоязнь и горизонтальный нистагм. В некоторых случаях отмечается появление врожденной глаукомы.
Причиной заболевания является нарушение функционирования гена PAX6 из короткого плеча 11 хромосомы. Кодируемый им белок приводит к запуску ряда процессов, которые управляют процессом правильного формирования органов зрения и ряда других структур. Примечательно, что ген очень консервативен: отличие форм PAX6 у человека и данио рерио составляет менее 5%, несмотря на расхождение эволюционных линий примерно 400 млн лет назад.
Заболевание относится к группе аутосомно-доминантных патологий. В случае гомозиготности по мутантной копии гена PAX6 негативный эффект на организм возрастает, что вызывает множественные нарушения в работе органов зрения. Кроме того, поражается ЦНС, что приводит к летальному исходу.
Лечение направлено на сглаживание симптомов. Для визуальной имитации зрачка рекомендуется использовать специальным образом окрашенные линзы. Возможно восстановление зрачка путем реконструктивной пластической операции.
Синдром Ди Джорджи
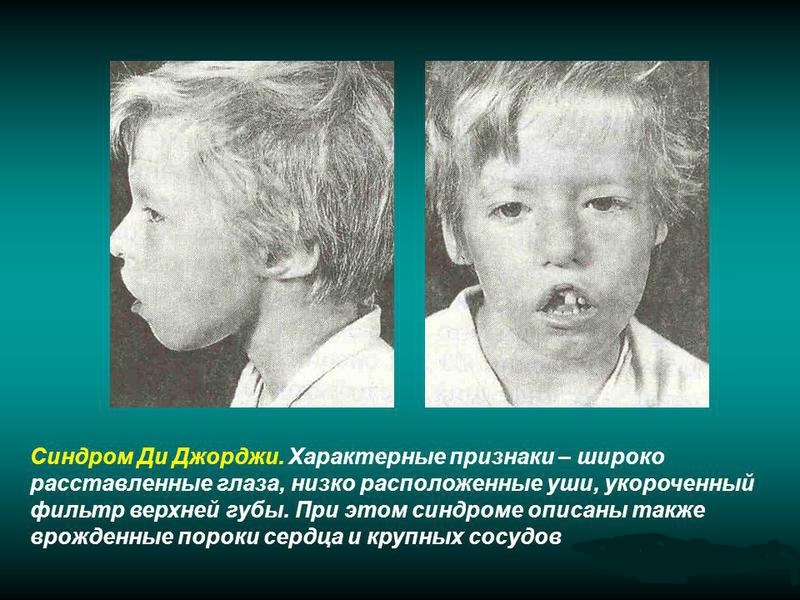
При синдроме Ди Джорджи у больных отмечается наличие врожденной формы аплазии паращитовидных желез и тимуса. Является разновидностью идиопатического изолированного гипопаратиреоза. Встречается достаточно редко.
При этом заболевании патологические изменения касаются околощитовидных (паращитовидных) желез, у которых отмечается дисгенез или агенезия. Вилочковая железа (тимус) отсутствует от рождения. В результате сочетания таких патологий происходит резкое снижение числа Т-лимфоцитов, формируется иммунологическая недостаточность. Кроме того, этот синдром сопровождается формированием врожденных аномалий крупных сосудов.
Заболевание является аутосомным и определяется наличие мутации в 22 хромосоме. В большинстве случаев причиной является спорадическая делеция 22q11 (реже микроделеция 22q11.2). Наследование происходит по доминантному принципу, с полом не связано. Некоторые авторы не соглашаются с такой характеристикой и приводят аргументы в пользу аутосомно-рецессивного типа, обладающего различной эспрессивностью.
Для заболевания характерно нарушение процесса эмбриогенеза 3-4 жаберных карманов, что приводит к нарушению закладки вилочковой железы и паращитовидных желез.
В клинике наиболее постоянными симптомами являются кандидомикоз и гипопаратиреоз, довольно часто сопровождающиеся нарушением процесса формирования рта, носа и ушей.
Тимус из-за нарушения развития в эмбриональном периоде остается неразвитым. Эпителий тимуса не обеспечивает нормального процесса развития Т-клеток. В итоге формируется специфическая форма иммунодефицита, при которой ослабляется гуморальный иммунный ответ и ответ на клеточном уровне. Если у ребенка имеется подобное патологическое нарушение иммунитета, то он будет обладать повышенной чувствительностью к инфекциям бактериального, вирусного и грибкового происхождения.
Синдром может протекать в форме генетически обусловленного отсутствия паращитовидных желез или изолированной недостаточности околощитовидных желез – в сопровождении гипокальциемических судорог, которые начинаются от рождения. Иммунологическая недостаточность приводит к появлению различных инфекционных заболеваний. Как правило, совокупность симптомов вызывает сердечную недостаточность. Кроме того, летальный исход вызывают инфекционные болезни.
Диагностика синдрома предполагает выявление типичных для синдрома патологий: искажения формы лица и черепа, наличие иммунологической недостаточности, аплазии тимуса, дисгенезии или агенезии паращитовидных желез. Ярче всего при заболевании проявляются кандидомикоз и гипопаратиреоз.
Ретинобластома
Ретинобластомой называют злокачественную опухоль сетчатки глаза. Процесс развития начинается обычно в детском возрасте, причем исходным материалом являются ткани эмбрионального происхождения. Пиковая фаза приходится на двухлетний возраст.
Практически все известные случаи выявляются в течение первых 5 лет жизни.
Причиной заболевания в большинстве случаев является мутация в генетическом материале. При этом необходимо наличие генетической обусловленности за счет наличия мутантной версии гена Rb, полученной по наследству. Вторая мутация, вызывающая появление опухоли, происходит в ретинобласте.
Существует небольшая вероятность, что у родителей, которые переболели ретинобластомой, могут родиться дети с отсутствием патологического изменения.
Отмечаются односторонние и двусторонние случаи ретинобластомы. По статистике для двусторонней формы вероятность наследственного происхождения заметно выше.
Симптомы заболевания включают боль в глазах, свечение зрачка, а также потерю зрения. Выявить их у маленького ребенка очень и очень трудно.
Диагностика обычно проходит в форме обследования под наркозом с применением УЗИ, КТ и МРТ. Достаточно распространенным приемом является биопсия красного костного мозга и спинномозговая пункция. По тяжести симптомов выделяется 5 групп.
Существует два эффективных метода лечения. При криотерапии и фотокоагуляции остается возможность сохранить и зрение, и сам глаз. Осложнения при их использовании возникают редко. Тем не менее, если возникнет рецидив, лечение потребуется повторить в той же форме. Обычно криотерапия используется в случаях, когда поврежден передний отдел сетчатки. Для заднего отдела более предпочтительным вариантом представляется фотокоагуляция.
Важность своевременной диагностики, в том числе пренатальной
Несмотря на минимальные изменения в генетическом материале, микроделеционные заболевания оказываются не менее опасными для здоровья человека, чем масштабные изменения. Поэтому важно вовремя определить наличие потенциально опасных мутаций. Благодаря пренатальной диагностике родители еще до момента родов могут убедиться, что у ребенка отсутствуют тяжелые генетические патологии. Постоянное совершенствование методов диагностики делает эти методы проверки все более эффективными и доступными.



