Альфа-Ритм
Медицинский консультативно-диагностический центр по вопросам диагностики и лечения эпилепсии и других пароксизмальных состояний Альфа-ритм
Телефоны для записи в Екатеринбурге
 +7-912-655-31-90; +7 (343) 287-55-05
+7-912-655-31-90; +7 (343) 287-55-05
Адрес в Екатеринбурге
Детские эпилептические энцефалопатии
ДЕТСКИЕ ЭПИЛЕПТИЧЕСКИЕ ЭНЦЕФАЛОПАТИИ С ПРОДОЛЖЕННОЙ СПАЙК-ВОЛНОВОЙ АКТИВНОСТЬЮ В МЕДЛЕННОВОЛНОВОМ СНЕ: КЛИНИЧЕСКАЯ ДИНАМИКА
Перунова Н.Ю., невролог-эпилептолог, д.м.н.
Детские эпилептические энцефалопатии с паттерном продолженной спайк-волновой активности во сне (CSWS, «continuous spike waves during slow wave sleep») принято рассматривать как модель воздействия эпилептиформной активности на высшие психические функции. Такие энцефалопатии, как эпилептический электрический статус медленноволнового сна (ESES, «electrical status epilepticus during slow wave sleep») и эпилептическая афазия Ландау-Клеффнера, считаются отдельными формами единого континуума.
Выраженность резидуального дефицита, формирующегося в исходе этих заболеваний, пропорциональна продолжительности активного периода. Критической длительностью активного периода эпилептических энцефалопатий с CSWS (ЭЭ с CSWS) предложено считать срок от 2 до 3 лет, после чего полноценное восстановление интеллектуальных функций невозможно.
В качестве отрицательных факторов прогноза ЭЭ с CSWS выделяют ранний возраст дебюта (до 3 лет), большую продолжительность активной фазы течения заболевания (более 3 лет), низкий уровень и длительное время диагностики, отсутствие ответа на терапию. Прогноз нарушений речи при эпилептических энцефалопатиях с CSWS считается неблагоприятным при дебюте заболевания в возрасте до 3 лет, сохранении речевых нарушений более 1 года.
В течении ЭЭ с CSWS предложено выделять активный период, характеризующийся персистированием паттерна CSWS, и резидуальный период. Проспективные исследования, содержащие информацию о динамике течения ЭЭ с CSWS, немногочисленны. Анализируются данные по группам из 11-18 пациентов (детей, подростков, молодых взрослых), длительность наблюдения от 5 до 9 лет, максимально – 16 лет.
Основными синдромами ЭЭ с CSWS являются наличие эпилептических припадков, расстройства поведения, когнитивные нарушения, нарушения речи, изменения на ЭЭГ.
Эпилептические припадки возникают у 80-90% пациентов, страдающих ЭЭ с CSWS. Назначение антиконвульсантов оказывается эффективным в отношении контроля припадков в 60% случаев, однако воздействие на эпилептиформную активность является недостаточным В отдаленном периоде эпилептические припадки могут сохраняться у 14% пациентов.

Рис.1. Сравнительная частота различных видов эпилептических припадков у пациентов с ЭЭ с CSWS (Перунова Н.Ю., Томенко Т.Р., 2004). ПП – простые парциальные; ВГСП – вторично-генерализованные судорожные припадки; АА – атипичные абсансы; ПАД. – эпилептические падения; СП – сложные парциальные припадки.

Рис.2. Фрагмент иктального паттерна парциального моторного (брахиофациального) припадка, развившегося во сне у пациента с ЭЭ с CSWS. Архив МКДЦ «Альфа-ритм».

Рис.3. Фрагмент иктального паттерна атипичного абсанса у пациента с ЭЭ с CSWS. Архив МКДЦ «Альфа-ритм».
Расстройства поведения в острой фазе ЭЭ с CSWS наблюдаются у 90% детей, однако при проспективном наблюдении у 75% из них отмечается постепенное снижение выраженности поведенческих нарушений.
Когнитивные нарушения в активном периоде ЭЭ с CSWS встречаются у 90% пациентов. Хорошее восстановление когнитивных функций возможно только у 10%, обучение в массовой школе – у 12%. Выраженный интеллектуальный дефицит в отдаленном периоде сохраняется у 18% подростков и молодых взрослых.
Полный регресс нарушений речи в исходе ЭЭ с CSWS возможен в 17-18% случаев, частичное восстановление нарушений речи — в 37-40%. У 50-60% пациентов речевые нарушения сохраняются в течение всей жизни.
Продолженная эпилептиформная активность с индексом более 50% — облигатный симптом ЭЭ с CSWS. В активном периоде эпилептиформная активность оценивается как диффузная в 40%, случаев, как диффузная с региональными преобладанием — в 60%. Клинические проявления ЭЭ с CSWS и характеристики эпилептиформной активности не всегда совпадают Персистирование изменений на ЭЭГ при LKS регистрируется до 5 лет, при ESES до 9 лет.

Рис.4. Фрагмент ЭЭГ сна пациента с ЭЭ с CSWS. Эпилептический электрический статус медленноволнового сна. Архив МКДЦ «Альфа-ритм».

Рис.5. Динамика основных синдромов ЭЭ с CSWS (собственная реконструкция по данным метаанализа).
К сожалению, доступные в настоящее время терапевтические воздействия на ЭЭ с CSWS недостаточно эффективны, что заставляет считать проблему ЭЭ с CSWS одной из наиболее актуальных и нерешенных в детской эпилептологии.
(Обзор составлен по данным публикаций: Smith MC. et al., 2003; Rossi PG. et al., 2004; Nickels K, Wirrell E., 2006; Tassinari CA, Rubboli G., 2006; Van Hirtum-Das M. et al., 2006; Wang S. et al., 2006; Praline J, et al., 2006; Scholtes FB. et al., 2010; García-Peñas JJ., 2010; Liukkonen E. et al., 2010; Loddenkemper T. et al., 2011.)
Эпилептическая энцефалопатия у детей

Эпилептическая энцефалопатия у детей – прогрессивное нарушение познавательных функций под влиянием эпилептического процесса. Проявляется дефицитом когнитивных, поведенческих функций, неврологическими отклонениями. Диагностика комплексная, включает клиническое обследование невролога и психиатра, энцефалографию, томографию, ультразвуковую допплерографию головного мозга, психодиагностическое исследование памяти, мышления, интеллекта, ряд лабораторных тестов. Лечение проводится противоэпилептическими препаратами, ноотропами, сосудорасширяющими, успокоительными средствами.
МКБ-10

Общие сведения
Термин «энцефалопатия» произошел из греческого языка, означает «болезнь, страдание головного мозга». Органическое повреждение его различных отделов при эпилепсии возникает вследствие интенсивных нейронных разрядов – чрезмерной биоэлектрической активности. Эпилептическая энцефалопатия (ЭЭ) обычно диагностируется у детей в первые месяцы жизни, распространенность в целом низкая, частота не превышает 0,05%. Эпидемиологические данные тяжелых синдромов искажаются ранней смертностью больных. Среди мальчиков заболеваемость стабильно выше – 60-66%. У дошкольников, подростков и взрослых ЭЭ развивается редко.
Причины
Эпилептическое органическое поражение головного мозга возникает как результат воздействия наследственных и анамнестических факторов. Причинами патологии являются:
К факторам риска развития ЭЭ у ребенка относятся патологические процессы при беременности (инфекции, интоксикации, травмы), осложненные роды, перенашивание и недонашивание плода, эпилепсия у ближайших родственников, травмы головы.
Патогенез
Прогрессирующее когнитивное и нейропсихологическое ухудшение при эпилептических энцефалопатиях у детей объясняется агрессивной иктальной и электрической эпилептогенной активностью в период созревания головного мозга. Ее интенсивность определяется стадией созревания мозга, возрастом дебюта. У новорожденных отклонения ЭЭГ представлены вспышкой-подавлением, у младенцев – гипсаритмией, у детей раннего возраста – генерализованными разрядами медленных волн.
Эпиактивность в левом полушарии отражается изменениями речевых функций – диагностируется афазия, аграфия, акалькулия, алексия, речевая диспраксия. Вовлечение в патологический процесс правого полушария проявляется агнозиями, монотонностью речи, бедностью жестикуляции, нарушениями артикуляции. Активность в орбитофрональных, цингулярных областях, срединных структурах формирует отклонения в поведении – аутистические, агрессивные проявления, мутизм. Патологические изменения эмоций, специфической памяти возникают при очагах в гиппокампальных структурах, амигдале.
Классификация
Эпилептические энцефалопатии у детей классифицируют по характеру течения и особенностям клинической картины. Выделяют два типа патологий – I и II.
Эпилептическая энцефалопатия I характеризуется прогрессирующими нарушениями речи, интеллекта, когнитивных и опорно-двигательных функций, отклонениями поведения, эмоций. Сопровождается эпилептическими приступами, развивается в рамках следующих синдромов:
При эпилептической энцефалопатии II определяются нарушения эмоционально-поведенческой, когнитивной сферы. Наблюдается быстрая утомляемость, агрессивность, сниженная работоспособность, трудности концентрации внимания. Эпилептические приступы отсутствуют.
Симптомы эпилептической энцефалопатии у детей
Клиническая картина у детей представлена спектром психических и поведенческих расстройств, нейропсихологическими нарушениями, свойственными органическим поражениям мозга при неврологических болезнях. Энцефалопатические признаки носят специфический характер, зависят от возраста больного ребенка. У детей первого года жизни наблюдается беспокойство, капризность, беспричинная смешливость или плаксивость. Реакции на изменение освещения, звука, положения бывают неадекватными. Дети часто срыгивают, неспособны сосать, плохо спят. Сердцебиение неравномерное, мышечный тонус повышен.
Дошкольники страдают нарушениями сна, интенсивными головными болями. Сохраняется высокий тонус мышц, развиваются обморочные состояния. Эмоционально-волевая сфера отличается неуверенностью, перепадами настроения, чертами гневливости. Дети быстро утомляются, имеют проблемы с запоминанием, трудности переключения внимания. Появляются первые признаки ригидности психической деятельности. В школьном возрасте полностью разворачивается симптоматика когнитивного дефицита. Определяется снижение функции запоминания, усвоения новой информации. Недостаточная гибкость мышления проявляется трудностями адаптации, интеллектуальным дефицитом. Сохраняются расстройства сознания, головные боли, головокружения. Формируется эпилептоидный тип личности с раздражительностью, рассеянностью, депрессивными состояниями.
Осложнения
Эпилептическая энцефалопатия у детей приводит к патологиям двигательной, физической, речевой и психической сферы. Прогрессирующие формы осложняются деменцией, олигофренией, гидроцефалией, детским церебральным параличом, психозами, аффективными расстройствами, психопатологическими изменениями личности. Дети не осваивают школьную программу, социально дезадаптированы, нуждаются в уходе со стороны. Ранняя диагностика и адекватное лечение снижают вероятность осложнений, пациенты способны обучаться в обычной школе, энцефалопатия завершается формированием минимальной мозговой дисфункции.
Диагностика
Диагностика эпилептической энцефалопатии у детей – трудоемкое комплексное обследование. Используются клинические, лабораторные и инструментальные методы:
Дифференциальная диагностика эпилептических энцефалопатий основана на результатах инструментальных исследований мозга. Характерно наличие эпиактивности, ее связь с клиническими проявлениями, улучшение состояния при приеме антиконвульсантов (для большинства форм заболеваний).
Лечение эпилептической энцефалопатии у детей
Лечение подбирается индивидуально, зависит от множества факторов – возраста ребенка, степени тяжести эпилепсии, причин ее развития. Специфическая терапия базируется на применении медикаментов. Используются следующие группы препаратов:
Дополнением к медикаментозной терапии является психокоррекция когнитивных функций, логопедические занятия, массаж, лечебная физическая культура. Комплексный подход позволяет купировать основные симптомы эпилепсии, устранить осложнения (речевые, интеллектуальные, двигательные).
Прогноз и профилактика
Прогноз определяется формой заболевания, своевременностью диагностики и адекватностью терапии. Наиболее благоприятный исход отмечается при эпилептической энцефалопатии II, эпилепсии с непрерывными спайк-волнами медленного сна: происходит ремиссия приступов, когнитивные способности улучшаются, поведенческие и эмоциональные нарушения ослабевают, достигается приемлемый уровень социальной адаптации.
Профилактика эпилептической энцефалопатии осложнена, так как заболевание развивается очень рано. Снизить вероятность эпилепсии позволяет тщательная подготовка и ведение беременности: расчет рисков генетических отклонений, своевременное лечение заболеваний будущей матери, отказ от курения, употребления алкоголя.
Эпилептические энцефалопатии
Эпилептические энцефалопатии составляют гетерогенную группу заболеваний, для которых характерны частые эпизоды судорог, устойчивость к лечению противоэпилептическими препаратами и существенная задержка психомоторного развития. Патология часто выявляется у детей разных возрастов, в том числе и у новорожденных. Среди наиболее актуальных проблем эпилептической энцефалопатии остается подбор специфической и эффективной терапии, которая смогла бы улучшить качество жизни пациентов.
Этиологические факторы
Причины развития ранней эпилептической энцефалопатии разнообразны. Их можно разделить на генетические и негенетические. К первой группе относятся:
К генетическим причинам эпилептической энцефалопатии относятся мутации в генах, которых насчитывается более 35: STXBP1, KCNQ2, CASK, SCN2A, SLC25A22, STK9, PLCB1 и т. д. Также заболевание может сочетаться с такими хромосомными аномалиями, как синдром Дауна или синдром Ангельмана. По типу наследования эпилептические энцефалопатии могут быть Х-сцепленными доминантными и рецессивными, а также аутосомно-доминантными и аутосомно-рецессивными.
Симптомы эпилептической энцефалопатии
Клиническая картина заболевания зависит от его тяжести, возраста ребенка, наличия или отсутствия дополнительных патологических изменений. Например, ранняя эпилептическая энцефалопатия может проявляться в виде неадекватной реакции ребенка на яркий свет или громкие звуки, повышенный тонус мышц, беспричинный плач и др. По мере взросления ребенка симптомы становятся более отчетливыми. Нарушения мышечного тонуса могут варьироваться от миоклонии и эпилептических спазмов до тяжелых судорог тонического или атонического типа.
В клинической картине обязательно присутствует задержка психомоторного или психоречевого развития, деменция, которая склонна к прогрессированию. Проявления заболевания могут дополняться очаговой неврологической симптоматикой.
Классификация эпилептических энцефалопатий
Классификация заболевания основана на клинике, этиологии, данных электроэнцефалографии и включает в себя 10 отдельных форм:
Данные формы заболевания могут переходить из одной в другую, поэтому специалисты озадачены доработкой классификации. Не исключено, что в ближайшее время она может измениться.
Диагностика эпилептических энцефалопатий
Сегодня существует большое количество методов, с помощью которых можно выявить ранние эпилептические энцефалопатии. Обследование начинается с общего осмотра и проверки рефлексов ребенка. Так как у новорожденных заболевание может сочетаться с метаболическими нарушениями, то дополнительно проводится исследование, направленное на их выявление. Это могут быть как генетические тесты, так и лабораторные анализы.
Характерную картину можно увидеть на электроэнцефалограмме. Для ранней эпилептической энцефалопатии характерно возникновение паттернов «вспышка-подавление», во время которых не прослеживается физиологический ритм. Этот признак обычно регистрируется во время сна, а в состоянии бодрствования может отсутствовать.
Для детей старшего возраста диагностика дополняется психологическими тестами, которые позволяют выявить расстройства психики, мышления, памяти, речи и других функций. Если требуется установить причину эпилептической энцефалопатии и особенности ее наследования, то врач назначит генетические тесты, направленные на поиск мутации в определенных генах. Пройти такое исследование можно в медико-генетическом центре «Геномед».
Эпилепсия в детском возрасте
Эпилепсия — общее название группы хронических пароксизмальных болезней головного мозга, проявляющихся повторными судорожными или другими (бессудорожными) стереотипными припадками, сопровождающихся разнообразными (патологическими) изменениями личности и сн
Часть 3. Начало статьи читайте в № 6, 8, 2014 год
Существует немало форм эпилепсии, встречающихся исключительно в детском или подростковом возрасте. Именно зависимость от возраста многих разновидностей эпилепсии является главным отличительным признаком эпилептологии детского возраста [1–4].
Эпилепсии и судорожные синдромы периода новорожденности
Хотя продолжительность неонатального периода невелика, целый ряд эпилептических синдромов свойственен именно для новорожденных детей [3–5].
Доброкачественные семейные приступы (судороги) новорожденных
Доброкачественная неонатальная эпилепсия (с аутосомно-доминатным типом наследования) трех типов, проявляющаяся в первые 7 дней жизни (начиная с трех суток). В семейном анамнезе обязательно фигурируют указания на наличие в прошлом судорог у членов семьи пациента (в неонатальном периоде). Связь припадков с уточненными врожденными нарушениями метаболизма не установлена. Доброкачественные семейные неонатальные приступы манифестируют в виде фокальных и мультифокальных или генерализованных тонико-клонических (судорожных) припадков. Указанные припадки характеризуются малой продолжительность (1–2 мин) и значительной частотой (20–30 эпизодов за сутки). Впоследствии, по прошествии от 1 до 3 недель, приступы самопроизвольно спонтанно купируются.
Доброкачественные несемейные судороги (приступы) новорожденных («припадки пятого дня»)
Эта эпилепсия с дебютом в раннем неонатальном периоде имеет также другое название (доброкачественные идиопатические неонатальные судороги). Болезнь впервые описана в конце 1970-х гг. Судорожные приступы развиваются у доношенных новорожденных детей, не имевших до этого признаков патологии со стороны ЦНС. Дебют приступов происходит к концу 1-й недели жизни (в 80–90% случаев — между 4-м и 6-м днями), а их пик приходится на 5-й день жизни (отсюда и название). Описываемые приступы обычно имеют вид мультифокальных клонических судорог, которым нередко сопутствуют апноэ. В большинстве случаев доброкачественные идиопатические неонатальные судороги длятся не более 24 ч (они всегда прекращаются по прошествии 15 дней после дебюта). В 80% случаев за время судорожного периода у новорожденных отмечается развитие эпилептического статуса [3–5].
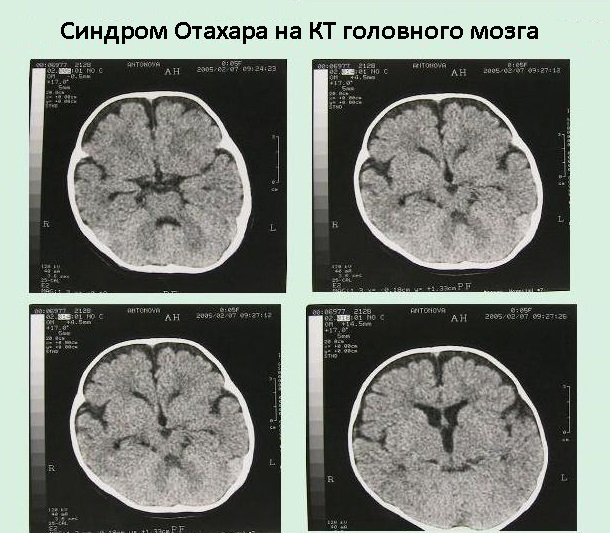
Ранняя инфантильная эпилептическая энцефалопатия с паттерном «угнетение/вспышка» на ЭЭГ (синдром Отахары)
Ранняя инфантильная эпилептическая энцефалопатия — редкая болезнь, относящаяся к злокачественным формам эпилепсии детского возраста. Дебютирует обычно в периоде новорожденности (или в возрасте 1–3 мес). Болезнь характеризуется тоническими приступами, частота которых значительно варьирует (10–300 эпизодов за сутки). У детей отмечается быстрое формирование неврологического дефицита и задержка психического развития. Специфический паттерн «вспышка/угнетение» при электроэнцефалографии (ЭЭГ) представлен у детей c синдромом Отахары как в состоянии сна, так и при бодрствовании. При магнитно-резонансной томографии (МРТ) головного мозга у пациентов отмечаются грубые аномалии развития ЦНС. Среди детей с ранней инфантильной эпилептической энцефалопатией с паттерном «вспышка/угнетение» на ЭЭГ летальность к возрасту 1 года достигает 40–50%. В 4–6-месячном возрасте синдром Отахары может трансформироваться в синдром Веста [3–6].
Ранняя миоклоническая (эпилептическая) энцефалопатия
Описана J. Aicardi и F. Goutières (1978); дебютирует преимущественно в периоде новорожденности (иногда до 3-месячного возраста). В генезе болезни предполагается роль генетических факторов и некоторых «врожденных ошибок метаболизма» (пропионовая ацидурия, метилмалоновая ацидемия, болезнь мочи с запахом кленового сиропа и др.). Клинически проявляется частыми миоклоническими припадками. Последние обычно не ассоциированы с ЭЭГ-изменениями во время приступа, но в ряде случаев одновременно с миоклониями регистрируются эпилептиформные разряды «угнетение/вспышка». Миоклонии чаще бывают фрагментарными (легкие подергивания дистальных отделов конечностей, век или углов рта); одновременно могут отмечаться фокальные (парциальные) приступы, массивные миоклонии и тонические спазмы (изолированные или серийные — возникают к 3–4 месяцам). Появление у ребенка тонических спазмов заставляет предположить наличие атипичного синдрома Веста, но вскоре основные проявления болезни возобновляются и сохраняются на протяжении длительного времени. Фокальные припадки (сложные парциальные — с заведением глаз или автономными симптомами: апноэ, гиперемия лица; клонические судороги в разных участках тела и др.) становятся основным типом приступов при ранней миоклонической эпилептической энцефалопатии. При интериктальном ЭЭГ-исследовании у детей регистрируется паттерн «угнетение/вспышка», состоящий из разрядов продолжительностью 1–5 сек, чередующийся с почти изоэлектрическими периодами (длительностью 3–10 сек). Описываемый ЭЭГ-паттерн становится более отчетливым во время сна (особенно в фазе глубокого сна). Изначальный паттерн «угнетение/вспышка» по достижении возраста 3–5 мес сменяется атипичной гипсаритмией или мультифокальными пароксизмами, но в большинстве случаев это лишь транзиторный феномен. Болезнь сопровождается высокой летальностью или прогрессивным распадом психомоторных функций (вплоть до вегетативного статуса), хотя по мере увеличения возраста частота и выраженность фокальных приступов и миоклоний постепенно уменьшаются [3–5, 7].
Витамин В6-зависимая эпилепсия
Cравнительно редкое наследственное заболевание, характеризующееся фармакорезистентными судорогами. Относится к группе метаболически обусловленных эпилепсий. Развивается у новорожденных, матери которых длительно получали пиридоксин во время беременности, а также при специфическом наследственном дефекте метаболизма (с повышенной потребностью в витамине В6). Известны случаи дебюта пиридоксинзависимых судорог у детей старше 1 мес и даже на втором году жизни. Между приступами судорог дети остаются беспокойными, реагируют мышечными подергиваниями на внешние раздражения. Болезнь не поддается обычному противосудорожному лечению, но назначение витамина В6 в высоких дозах (25 мг/кг/сут) быстро приводит к нормализации состояния [3–5].
Злокачественные мигрирующие парциальные судороги (приступы) младенческого возраста
Чрезвычайно редко встречающийся эпилептический синдром, описанный G. Coppola и соавт. (1995). К настоящему времени сообщается всего о примерно 50 случаях болезни, зарегистрированных в различных странах мира. Злокачественные мигрирующие парциальные судороги в 50% случаев наблюдаются в первые дни жизни; остальные 50% приходятся на возраст 1–3 мес. При дебюте приступы носят фокальный клонический характер, а по прошествии нескольких недель они становятся мультифокальными, причем исключительно частыми и фармакорезистентными к терапии антиэпилептическими препаратами. При ЭЭГ-исследовании у детей выявляется выраженная многоочаговая эпилептическая активность; метаболических нарушений не обнаруживается, а МРТ-признаки патологических изменений отсутствуют. Паталогоанатомическое исследование позволило выявить в гиппокампе признаки нейрональной потери [1, 3, 5, 8].
Эпилепсии у детей первого года жизни (1–12 мес)
По достижении 1-месячного возраста число разновидностей эпилептических синдромов, специфичных для первого года жизни ребенка, практически не уступает таковому, свойственному периоду новорожденности.
Инфантильные спазмы (синдром Веста)
Этот вариант катастрофической эпилепсии (генерализованной) бывает симптоматическим (подавляющее большинство случаев) или криптогенным (10–20%). Он манифестирует у детей на первом году жизни (чаще между 3-м и 8-м месяцами). В классическом варианте синдром Веста характеризуется в момент приступа комбинацией сгибательных и разгибательных движений, то есть выраженными миоклоническими (салаамовыми) спазмами, иногда серийными короткими сгибательными движениями головы («кивки»). Инфантильные спазмы могут развиться как вследствие наличия различной неврологической патологии, так и без каких-либо очевидных предшествующих нарушений функций ЦНС. При инфантильных спазмах психомоторное развитие замедляется, в дальнейшем высока вероятность выраженного отставания в развитии. В 80% случаев при синдроме Веста обнаруживаются микроцефалия, признаки детского церебрального паралича, атонически-атактические нарушения и др. Отличительным электрофизиологическим признаком синдрома Веста является гипсаритмия (по данным ЭЭГ), которая имеет вид диффузных высоковольтажных пиков и медленных волн, располагающихся на дезорганизованном (медленном) фоне. Прогноз синдрома Веста определяется эффективностью проводимой терапии, но в целом малоблагоприятен [3–8].
Тяжелая миоклонус-эпилепсия младенческого возраста (синдром Драве)
Болезнь, описанная C. Dravet (1978, 1992), дебютирует на первом году жизни (между 2-м и 9-м мес), что нередко происходит вслед за развитием фебрильного эпизода, вскоре после вакцинации или перенесения инфекции. Синдром Драве характеризуется появлением генерализованных или односторонних клонических судорог (обычно на фоне гипертермии или лихорадки), что происходит на фоне предшествующего нормального психомоторного развития ребенка на протяжении первого года жизни. Постепенно (по прошествии нескольких недель или месяцев) у ребенка развиваются афебрильные миоклонические и парциальные (фокальные) припадки. Прогрессивное нарастание частоты миоклоний (изолированных или серийных) предшествует появлению у пациентов генерализованных припадков. У детей выявляются умеренные мозжечковые и пирамидные знаки, связанные с дефицитарностью грубой моторики и атаксией походки. Нарушения психомоторного развития впоследствии отмечаются у детей примерно до 4-летнего возраста. Нередко при синдроме Драве у детей развивается эпилептический статус (судорожный или миоклонический). Данные ЭЭГ на протяжении первого года жизни обычно остаются в пределах нормы, хотя у отдельных пациентов встречаются спонтанные фотоиндуцированные пик-волновые разряды. Впоследствии иктальные ЭЭГ-исследования при синдроме Драве характеризуются наличием миоклонических или клонических припадков (генерализованная пик-волновая или полипик-волновая активность). Генерализованные разряды усиливаются в состоянии релаксации; одновременно отмечаются фокальные и мультифокальные пики и острые волны. Традиционные и новые антиэпилептические препараты обычно не предотвращают рецидива приступов при синдроме Драве. Прогноз по интеллектуальному развитию при синдроме Драве всегда неблагоприятен [3–5, 8].
Идиопатические доброкачественные парциальные эпилепсии младенчества
Обычно дебютируют у детей в возрасте 3–20 месяцев (чаще между 5-м и 8-м мес). Впервые описаны K. Watanabe и соавт. (1987), вследствие чего изначально получили обобщающее название «синдром Ватанабе». Характеризуются проявлениями в виде сложных парциальных (фокальных) приступов и благоприятным прогнозом (элиминация эпилептических припадков в течение 3 мес после дебюта). В среднем число приступов составляет около 7; у части пациентов отмечаются исключительно сложные парциальные припадки, у других — только вторично-генерализованные, а примерно в половине случаев встречается их сочетание. Во время приступа для пациентов характерны снижение реакции на предъявляемые стимулы, остановка двигательной активности, умеренные судорожные подергивания, латеральное заведение глаз и цианоз. Основными клиническими признаками этой группы эпилепсий являются высокая встречаемость кластерных приступов, короткая продолжительность припадков, а также изначально нормальные показатели интериктального ЭЭГ-исследования (впоследствии у части детей могут обнаруживаться пароксизмальные разряды) [2, 3, 5, 6, 8].
Сходные с идиопатическими доброкачественными парциальными эпилепсиями младенчества, но исключительно семейные пароксизмальные состояния с дебютом на первом году жизни носят название «доброкачественные инфантильные семейные судороги». В 1997 г. были описаны сходные с ними случаи семейных эпилепсий с последующим формированием хореоатетоза — семейные судороги с хореатетозом [3–5, 8, 9].
Эпилепсии у детей раннего возраста (1–3 года)
Для детей раннего возраста (от 12 до 36 месяцев), в первую очередь, характерны cиндром Доозе, синдром Леннокса–Гасто, доброкачественная миоклонуc-эпилепсия младенческого возраста, синдром гемиконвульсий-гемиплегии, идиопатическая парциальная эпилепсия младенчества, абсансная эпилепсия раннего детства, электрический эпилептический статус медленно-волнового сна, ранний и поздний детский нейрональный липофусциноз (типы I и II).
Миоклоническая астатическая эпилепсия раннего детского возраста (cиндром Доозе)
Представляет собой эпилепсию c миоклонически-астатическими приступами (различной продолжительности). Приступы дебютируют в возрасте 1–5 лет. Чаще болезнь поражает мальчиков. Астатические и миоклонические приступы могут сочетаться, причем миоклонии возникают как до, во время, так и после астатического припадка. Приступы наступают внезапно и практически всегда сопровождаются падениями. Миоклонии отмечаются в виде различной выраженности симметричных подергиваний в руках и мышцах плеч пояса, что сочетается с наклоном головы («кивки»). Признаки утраты сознания у детей в момент приступа отсутствуют. До начала заболевания психомоторное развитие детей обычно соответствует норме. У части детей болезнь осложняется риском развития деменции (предположительно за счет развития эпилептического статуса абсансов). При ЭЭГ регистрируются генерализованные билатерально-синхронные комплексы пик-волн (3 и более за 1 сек, 2–4 Гц). Прогноз при миоклонически-астатической эпилепсии раннего детского возраста малоблагоприятен [3–6, 8].
Синдром Леннокса–Гасто, или миокинетическая эпилепсия раннего детства с медленными пик-волнами
Группа гетерогенной патологии с эпилептическими приступами (атоническими, тоническими, атипичными абсансами), интеллектуальной дефицитарностью и характерным ЭЭГ-паттерном. Как и при синдроме Веста, при синдроме Леннокса–Гасто выделяют симптоматический и криптогенный варианты болезни. Ранние формы дебютируют примерно с 2-летнего возраста. До 30% случаев представляют собой результат трансформации из синдрома Веста. Клинически синдром Леннокса–Гасто характеризуется миоклонически-астатическими припадками, салаамовыми судорогами (молниеносными кивательными), атипичными абсансами, тоническими приступами (чаще во сне). Могут встречаться генерализованные тонико-клонические, миоклонические и фокальные (парциальные) приступы. Для детей типична серийность припадков с изменениями сознания (ступор) и постепенным переходом в эпилептический статус. Помимо эпилептических приступов, в неврологическом статусе могут отмечаться церебральные парезы/параличи, а также атонически-астатические нарушения (до 40% пациентов). У детей происходит снижение интеллекта (различной степени), наблюдаются выраженные нарушения когнитивных функций. По ЭЭГ-данным типичны изменения фоновой активности в виде медленных пик-волн
В. М. Студеникин, доктор медицинских наук, профессор, академик РАЕ
ФГБУ «НЦЗД» РАМН, Москва



